质谱检测法与蛋白质分析(三)

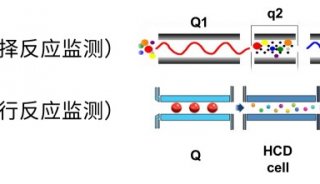
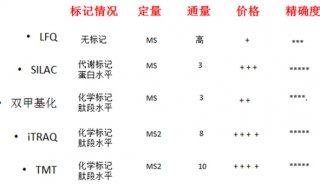
Protein(s):待测蛋白质样品;Enz. Digestion:酶解;Pep. Mixture:裂解产物混合物;
MS Analysis:质谱检测分析;DB Search:数据库比对搜索;Identities:鉴定;
Prot.DB :蛋白质数据库;Proteome:蛋白质组;Sample prep:样品准备(裂解);
LC/MS& MS/MS:LC/MS以及 MS/MS分析;Quantification:定量研究;
Abundances:丰度;List of targets:挑选出待测样品;Pep.lib:待测靶样品;
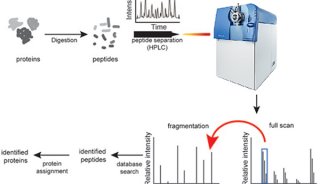
图 2 蛋白质组学研究策略示意图。 A :经由 2 维电泳或 pull-down 实验发现的待测蛋白质样品 prot 随即被酶解,然后用质谱仪对酶解片段 pep 进行质谱检测分析。最后根据肽质指纹图谱( PMF )鉴定出肽段以及它们的来源蛋白。其它的 MS/MS 数据也能用来进行肽段鉴定。 B :随机蛋白质鉴定和定量分析方法,即鸟枪法。该方法可以对样品同时进行鉴定以及定量研究。被选出的肽段如图 1A 中所示,经过串联质谱仪进行离子扫描分析。在这里,母离子是被随机选出的,通常母离子的裂解片段中只有一个片段能够被检测到,也就是说存在采样不足的问题。 MS1 质谱仪采集到的离子强度信息与参考分子信息比对可以进行定量分析,这里使用的参考分子通常都会标记上稳定的同位素标签。 C :定量研究方法能去除蛋白质定量研究与鉴定过程中的干扰信号。首先, MS1 质谱仪会通过比对不同样品的肽模式,即肽段分子量相对色谱保留时间作图的结果,挑选出不同样品间丰度不同的肽段。然后被挑出的肽段进入下一轮 MS/MS 质谱检测。 D :基于各种假说的检测方法。该方法可以对各种预先被设定肽段的丰度进行高精度的检测,我们通常都是根据以往的实验结果来挑选这些靶肽段。在这里常用的方法是图 1D 中所示的 MRM 方法。如果在试验中选用合适的参考肽段会进一步提高实验的准确性。
比较模式分析
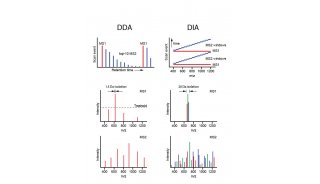
图2C中所示的比较模式分析法其实在原理上与2维电泳方法是相似的,就像在2维电泳中一样,每一个蛋白质都是一个独立的点,可以借此对它们进行定量和定性的分析。而且还可以进一步对蛋白质进行分析,比如进行蛋白质测序或进行翻译后修饰情况鉴定等。不过在以质谱检测手段为基础的分析方法中,蛋白质样品首先需要被降解、片段化,然后再用液相色谱-质谱仪LC/MS进行分析。借助色谱洗脱时间和分子量这两个参数,我们就可以鉴定出一个多肽离子。将所有离子的数量信息整合起来就得到了蛋白质的定量信息。这种方法主要的优势在于它可以对质谱仪发现的所有信息进行定量处理。与“鸟枪法”相比这是非常明显的优势,因为在“鸟枪法”里,只能对被鉴定出来的肽段进行定量化处理。不过在实际运用过程中,这种比较模式分析方法的重复性很差,也没有非常好的配套软件对检测结果进行分析。用这种方法可以获得一些数据,根据这些数据能够推测出蛋白质序列,这些数据包括质荷比(m/z)、带电荷状态、洗脱时间以及离子强度等。需要被测序的多肽(比如在两个样品中表达丰度不同的同一蛋白质)会出现在检测结果列表中,然后,我们会将该蛋白(多肽)进行新一轮的直接质谱检测,专门只采集它的MS/MS质谱图信息。这种研究方法也可用于MALDI/MS/MS质谱检测平台,因为蛋白质样品都被“固定”在样品板上,因此可以对它们进行不受时间限制的连续检测。
基于各种假说的研究策略
随着各种质谱仪技术的不断进步,我们有理由相信会出现敏感度更高、处理速度更快、更可靠的蛋白质组研究设备。不过目前我们还不清楚这些技术上的进步是否足以突破当下蛋白质组学研究工作中的瓶颈。以前我们曾认为蛋白质组学研究策略需要进行有效的变革才能全面、有力的对蛋白质组进行研究和分析。这种变革的核心就是改变以往那种在每一次试验中都对既往结果进行重复研究的蛋白质组学研究方法,新的研究策略是根据既往的研究成果来设计、指导进行新的实验研究项目。我们预计与人类基因组计划一样,将来也会获得某个物种的完整蛋白质组序列图谱,未来蛋白质组的研究目标应该是对蛋白质(多肽)进行具有特定目的的、非冗余的分析研究。对于质谱技术和质谱仪器的发展来说,这种研究策略的转变需要有更好的检测设备和检测手段(流程)做依托,要能够以更快的速度、更高的灵敏度和更强大的分析能力进行质谱检测。现在也出现了可供序列组装、修饰情况查询的数据库。可以预计,在不久的将来,各种生物学假说将会给我们设定出许多待检测的蛋白质靶标。我们可以将这些蛋白质的信息录入数据库,构成一个检测这些假说所必需的极小集蛋白质信息簇。然后,可以用各种方法,比如MRM方法对这些蛋白质进行检测(图2D)。因为这种研究方法主要是对非冗余靶标进行质谱检测,所以,在检测的敏感度方面和样品处理能力方面都有很大的提高。如果再配合定量的、标准化的同位素标记参考肽段,还可以进行精确的定量分析。
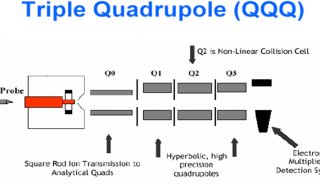
上述这种研究策略最适合用于Q-Q-LIT质谱仪这类被我们使用了几十年,主要用于检测小分子药品和体内药物代谢研究领域的三重四级杆质谱仪。在研究药物代谢时使用的研究方法同样适用于蛋白质组学研究领域。
作为上述方法的一个补充,Smith又开发出另一种使用精确分子量标签来鉴定蛋白质的方法。该方法首先测定待测物质的分子量,然后将数据与分子量数据库进行比对,以此来鉴定蛋白质。这样就无需再对每一个样品中的每一条多肽进行测序了。随着蛋白质组学研究的不断开展,我们积累的数据也越来越多,并且数据积累的速度也越来越快,同时,各种分析软件的功能也越来越强大,因此我们相信,基于各种假说的蛋白质组学研究策略一定会被更多的人所接受,所采用。

Labeling:标记 Tag:标记物 Peptide:肽段 Mixing:混合 Analysis:分析
MS:质谱检测 light:较轻的片段 heavy:较重的片段 Time:时间
Tandem mass tag:串联标签 MS/MS:MS/MS模式 m/z:质荷比
No labeling!:为标记 MRM:MRM方法
Internal standards(isotopically labeled peptides):内参,即同位素标记的肽段
图 3 肽段定量分析策略示意图。 A :同位素稀释法是进行肽段定量分析时最常用的一种方法,该方法也是进行蛋白质组学研究时常用的一个方法。该方法的原理是在一号样品所有的待测蛋白质上都带上一个稳定的同位素标签,同时在二号样品所有的待测蛋白质上都带上另一个稳定的同位素标签。这样,这两组样品就可以互为参照了。可以借助化学的方法,例如稳定的同位素编码标签试剂、代谢标记反应和酶标反应等对蛋白质进行同位素标记。 B :使用串联标签进行定量研究。该方法同样需要各种稳定的同位素标记物。这些同位素标记物由两种同位素标记元素组成,它们都有固定的分子量。目前,这些试剂可用于四通道反应。通过在质谱仪的 MS/MS 模式下检测连接在蛋白质 N 端报告基团的相对强度以及 CID 图谱中低分子量范围里的信号可以获得待测蛋白质的定量信息。 C :使用内参进行定量研究的方法。该方法实际上也是一种同位素稀释法。在该方法中会往待测样品中添加一种已知浓度的同位素标记的肽段,然后借助标准曲线进行精确的定量分析。虽然该方法样品制备过程较为复杂,但是它的前景非常光明。它非常适合用于基于各种假说的检测方法。
结论与展望
在过去的十年里,是蛋白质分析,更准确的说应该是蛋白质组学分析推动了质谱检测技术的发展。各种技术进步已经使质谱仪在准确度、分辨力、敏感度、定量分析能力等各方面都有了长足的进展,蛋白质组质谱检测策略方面也有了新突破。分析完整蛋白质、蛋白质复合体、低丰度蛋白质等各种样品的检测操作流程层出不穷。虽然这些质谱检测技术是为了满足蛋白质分析的需求而诞生的,但是它们出现之后又反过来推动了生物大分子(包括代谢产物、脂类物质、碳水化合物等等)领域的研究。因此,我们有理由相信,质谱检测技术必将在生物研究的各个领域里占有重要的一席之地。
-
焦点事件

-
标准

-
企业风采