基于 Orbitrap GC-MS 的非靶向代谢组学(三)
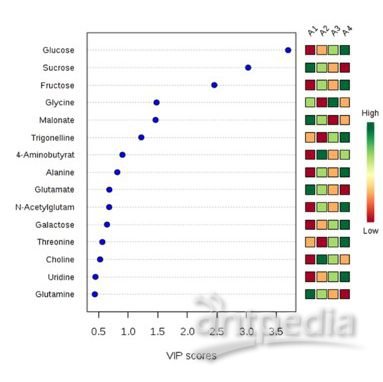
使用 SIMCA 软件进行多变量统计分析。5将结论数据转化为更为直观的对数值,将 Y 类别设为各时间点,生成数据的偏最小二乘判别分析(PLS-DA)模型。该软件生成分数图和载荷图,以显示沿着主成分每个时间点的聚类和分离(图 6)情况。从本次分析可知,死亡后立即取样的样品(RAT_T0)聚集在一起,与已分解的大鼠样品(T1–T3)具有显著性差异。从 T1–T3 样品中可以观察到分组聚类和持续分解。X 或 Y 轴上的位移表示某一代谢物对分数图中样品群组之间分离的作用大小。在本例中,X 轴将 T0 与 T1–3 分开,Y 轴将 T1、T2 和 T3 分开(图 6)。载荷图上的每个蓝点表示每个已检测到的含 EI 碎片离子簇的代谢物(图 7)。
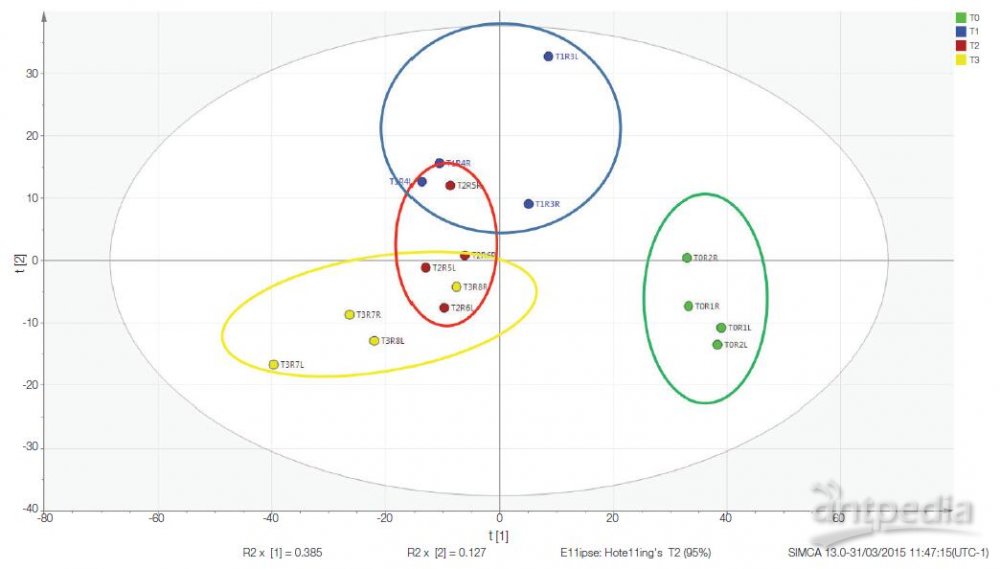
图 6. 分解数据的 PLS-DA 模型。该模型为有监督多变量分析,将高维数据(例如,大量强度不同的代谢物)折叠为涵盖数据集中主要变化的几种主成分。在本例中,X 轴是主成分 1,Y 轴是主成分 2。注意,本项研究对样品进行了适当聚类,每个群组聚集在一起,T0 已明显与其他群组分开。

图 7. PLS-DA 模型的载荷图。根据代谢物(用蓝色原点表示)对群组分离的作用大小对代谢物进行聚类分析,见图 6。例如,最右边的代谢物对于定义 T0 样品作用很大。
表 4. 分解期间含量不断增加的代谢物的推定 ID 列表。可以通过精确质量数和分子式推测进行化合物确认。只有在高分辨率和高质量数精度条件下才能检测到腐胺,并将其从背景离子中解卷积出来。
推测化合物 ID | RT(min) | NIST 正向匹配 | 与 T0 相比 的增加倍数 | 基峰碎片 元素组成 | ppm 精度 (基峰) | ppm 精度 (分子离子) |
L 苏氨酸,3TMS | 10.71 | 795 | 2.8 | C9H24ONSi2 | 0.27 | 0.13 |
L 天冬氨酸,3TMS | 11.78 | 707 | 7.0 | C9H22NO2Si2 | 0.18 | 0.34 |
L 甲硫氨酸,2TMS | 12.40 | 749 | 15.0 | C7H18NSSi | 0.24 | 0.04 |
L 谷氨酰胺-3TMS | 15.32 | 815 | 2.0 | C7H14NOSi | 0.53 | 0.21 |
腐胺,4TMS | 16.18 | 870 | 2.0 | C7H20NSi2 | 0.05 | N/A |
赖氨酸,4TMS | 16.88 | 732 | 5.1 | C8H18NSi | 0.19 | 0.05 |
识别阶段
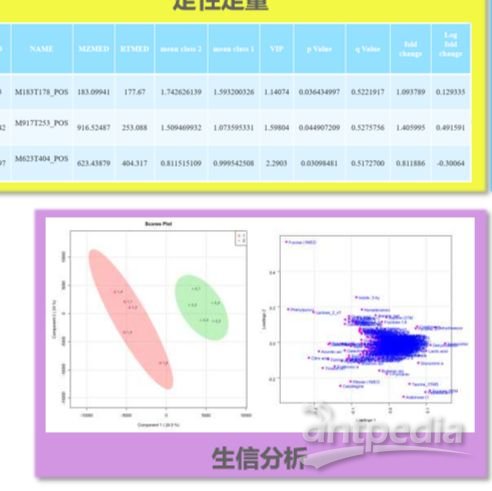
依据现有商用库(NIST)识别发生显著性变化的代谢物,首先将其与氨基酸、与分解相关的化合物进行匹配(表 4)。基于多变量统计分析的整个流程总结见图 5,图中对峰进行了解卷积、定量和识别。所有选择的化合物得到的分数均较高(> 700),使用精确质量数进行碎片匹配,从而进一步提高了库匹配结果的准确度(表 3)。
结论
在此介绍的 Q Exactive GC 系统的工作流程顺序为样品制备、自动衍生化、GC 分离和质谱检测、数据分析和结果报告。这一工作流程使 Q Exactive GC 系统成为挥发性化合物和需衍生化非挥发性化合物进行代谢组学分析的独特分析工具。
卓越的色谱分离能力、可重现的色谱分离结合快速数据采集使 Q Exactive GC 系统成为复杂代谢组学分析的理想平台。
超高分辨率、始终如一的亚 ppm 的精确质量数测量为复杂生物分解基质中存在的多种代谢物提供了可靠和高选择性的分析。
较宽的动态范围为分析样品中的代谢物提供高灵敏度和持续检测,且质量数精度丝毫不受影响,同时为已检测到的代谢物提供精确的相对定量。
可依据已有商用库使用获得的 EI 数据对化合物进行初步识别,使研究人员对结果进行评价。同时,得到的精确质量数允许对数据进一步分析,如使用裂解分析(例如,Mass Frontier)或使AN 10457_C_GCMSMS_201508Y用标准品进一步确认目标化合物。本例中,氨基酸信号的时间依赖性演变为检测死亡时间提供了一种简便的生化法医分析检测法。
参考文献
1. ProteoWizard Page. http://proteowizard.sourceforge.net/
(accessed Apr. 22, 2015).
2. C.A. Smith, E.J. Want, G.C. Tong, R. Abagyan, and G. Siuzdak. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem.
2006, 78 (3), 779–787.
3. Richard A. Scheltema, Andris Jankevics, Ritsert C. Jansen, Morris A. Swertz, and Rainer Breitling. PeakML/mzMatch: A File Format, Java Library, R Library, and Tool-Chain for Mass Spectrometry Data Analysis. Anal. Chem.
2011 83 (7), 2786–2793.
4. Creek, D.J., Jankevics, A., Burgess, K.E.V., Breitling, R., and Barrett, M.P. IDEOM: an excel interface for analysis of LC-MS based metabolomics data. Bioinformatics.
2012. 28 (7), 1048–1049.
5. SIMCA Page, Umetrics Website. http://www.umetrics. com/products/simca
(accessed Apr. 22, 2015).