新药IND申请中的CMC申报
药品的化学、制造和控制,简称CMC,是产品成功开发并注册上市的关键要素之一,CMC申报是药品申报资料中非常重要的部分。IND中CMC的信息一般包括原料药、制剂的组成、生产工艺、稳定性和生产控制、安慰剂的信息(特别是在临床期)、药品的外标签、环境评估信息,其目的是充分证明生产商可以连续生产和提供质量稳定和统一的测试样品。
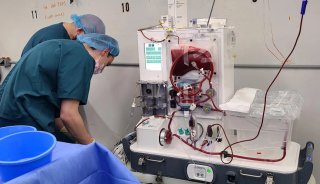
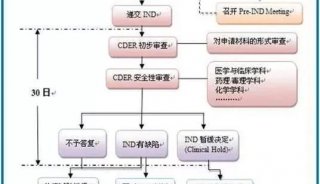
美迪西药物分析提供全方位的药物分析服务,包括方法开发和方法验证,分析测试与放行,稳定性研究,大规模分离和CMC申报文件服务。FDA对IND中CMC的信息作了限定要求:首先,应有足够的资料证明应试药物的性状、质量、纯度和剂量;其次在IND中需要有一个部分用以描述原料药和制剂的组成,生产和质量控制。在IND的不同周期,需要提交的IND资料是不一样的。
自2018年5月以后,所有到美国申报的IND需要遵循eCTD(电子通用技术文档)要求,eCTD对CMC信息的要求主要涉及资料格式和结构。
1、提交足够的CMC资料
在CMC资料够与不够这个问题上,前FDA CMC审评专家、Advaxis公司法规事务监管杜新博士认为很难有明确的共识,如果将IND的CMC资料递交给两个FDA审评员,他们的意见也可能不统一。提供的CMC资料是否足够要考虑以下因素:
(1)IND在期还是期,是否含有未知或不纯的成分;
(2)产品是否含有已知或极可能有毒性的化学结构,毒性高低与否;
(3)产品在测试项目过程中是否能保持化学稳定性;
(4)产品是否分布有杂质或有潜在的危害健康因素;
(5)是否有杂质分布无法被有效检测出导致无法评估其潜在的健康危险,以及不适当的标签。
对这些做充分分析理解后,或许就明白CMC信息究竟多少是够的,另外,杜博士提到FDA有关Cross-reference(交叉引用),FDA将一个indication(适用症)为一个IND,不同的indication在临床报的时候IND可以不一样,但药是一样的,其中包含的CMC信息文件是一样的。 因此, 在进行下一个IND时无需提供CMC信息文件,只需要Cross-reference。
2、导致CMC暂停潜在的问题
导致CMC暂停的原因主要从以下几方面分析考虑:
(1)证明药品安全性的信息不足,存在较高毒性隐患;
(2)临床试验批次杂质不合格或杂质信息表征不充分;
(3)主细胞或工作细胞库或病毒库的研究不充分;
(4)药品在试验过程中不稳定;
(5)毒理学研究未定义;
(6)非临床试验数据不能支持最大人体剂量的界定;
(7)临床方案设计不符合规定的目标;
(8)临床研究设计缺乏必要的安全监测,以及/或“试验中止规则”未被定义或不充分;
(9)非临床批次与临床批次之间缺乏关联性。
3、CMC申报中存在的常见问题
(1)原料药中常见问题 :杂质未定义明确,包括工艺杂质、制剂杂质、容器中浸出杂质或残留溶剂等;对原料药的了解不够充分;遗传毒性研究不充分;缺乏药物与安全相关的临床与临床前关联评价;方法及放行标准不适用。
(2)制剂中常见问题:方法及放行标准不适用;处方组成中辅料的了解程度不够;没有足够的稳定性数据支持临床周期;设备适用性问题;包材相容性问题。
(3)辅料:IND中对辅料缺乏说明;与药物的相容性问题;标准问题(USP/NF;FDA非活性成分数据库);用CP标准可能会存在问题;供应商信息(期以后,供应商改变后标准有没有变);创新辅料的问题;动物型辅料来源问题(如疯牛病风险)。
(4)安慰剂:重视程度不够;CTD中问题,信息可以较制剂产品相对少,但关键信息不能缺。
(5)生物药产品中常见问题:生物药产品相对小分子药来说结构和分子量比小分子大得多,怎样确定其结构、纯度、效和安全性的问题;生产需求条件更严苛—GMP认证;缺乏病毒清除;高风险产品缺乏或不适当的免疫原性测定;缺乏最终药品无菌证据;缺乏必要的安全评估。
大分子和小分子新药申报的CMC的相同点是:都需要符合IND要求,遵循eCTD格式,让审评员能轻松审阅理解。其不同点如下表所示:
小分子 | 大分子 | |
分子大小 | 小 | 大 |
结构复杂程度 | 简单 | 复杂 |
IND/CTD中内容 | 较少 | 较多 |
遗传毒性评估 | 有 | 多数无 |
制剂处方 | 相对复杂 | 简单(注射剂) |
生产要求 | 简单 | 复杂 |
设备要求 | 简单 | 需满足GMP要求 |
溶解性 | 测定 | —— |
免疫原性 | 无 | 有 |
储运条件 | 简单 | 复杂严苛 |
大分子和小分子新药申报的CMC的不同点
4、临床试验注册中的CMC的关键点
(1)、期临床试验是否必须要要cGMP。
(2)、期临床产品是不需要按照 21 CFR 211上市产品要求,实际上21 CFR 211是主要针对的是商业产品而不是期药品,不过还是要按照“缩减版”的cGMP要求。具体有三点:要有明确的操作规程;能够充分控制的设备和生产环境;要准确、同步记录生产中(包括测试)的数据。
(3)申报IND需要多少个批次批量及稳定性数据
至少两个批次,如毒性试验的批次和临床试验的批次;批量只要是代表工艺实际就可以;稳定性数据最冒险的是提交两个点,也就是0和1个月的数据。随着资料的整理,最后递交的时候补上3个月的稳定性数据即可。
(4)建立药物的杂质库
在临床试验注册中,还应该建立药物的杂质库,尽可能明确定义杂质,对于未知杂质使用ICH指导原则予以限定,校验杂质残留的耐受程度,使用合适的测试方法和适当的标准,对自身产品进行充分的鉴定评估。