Nature Methods | 郑晔开发三维基因组数据高保真模拟新工具
染色体构象捕获3C(chromosome conformation capture【1】)和Hi-C(high-throughput chromosomeconformation capture【2,3】)测序技术已经逐渐成熟,成为研究远距离基因调控机制的重要方法。越来越多的学者提出分析Hi-C数据信号探测的模型和算法,但由于Hi-C数据深度测序成本巨大,而大部分检验结果无法进行实验验证真伪,因而迫切需要一个稳定的Hi-C数据模拟方法来进行基准测试和评估。目前,只有一个系统的Hi-C 数据模拟方法即Sim3C【4】,却由于三维基因组数据结构过于复杂,Sim3C模型无法捕获所有Hi-C数据的基本特征,再加上参数模型强加的一系列假设,使得模拟出来的Hi-C数据矩阵与真实Hi-C数据矩阵相差甚远(下图)。此外降采样(downsampling)和生物样本间置换(permutation)作为常见的测试方法,也有各自无法避免的局限。

2019年11月11日,来自威斯康星大学麦迪逊分校的郑晔博士和 Sunduz Keles教授在Nature Methods发表了题为“FreeHi-C simulates high fidelity Hi-C data for benchmarking and dataaugmentation”的论文,提出了一个全新的Hi-C数据高保真模拟方法,即FreeHi-C, 使得三维基因学基准测试更为合理,并且通过模拟数据扩增(data augmentation)显著提高了信号差异分析的检测精度(precision)与强度(power)并且有效地控制错误发现率(false discovery rate)。

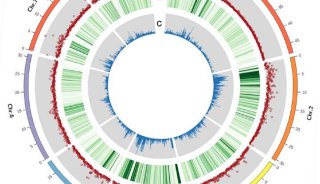
目前,Hi-C数据的模拟方法都是通过学习Hi-C数据矩阵特征,比如接触距离(contact distance)、拓扑相关结构域(TopologicallyAssociating Domains,TADs)、染色质环( chromatin loops )等,直接模拟出n x n的矩阵。但由于基因组三维立体结构非常复杂,结构之间的关系也尚未明晰,因而直接模拟矩阵效果并不理想。FreeHi-C则选择从Hi-C实验过程【2,3】的角度,通过非参数模型,模拟实验中每一步生成的数据,获得模拟基因序列(sequencing data),再进行与真实数据相同的基因序列处理,得到高保真的Hi-C模拟数据矩阵(下图)。FreeHi-C一大优势是使用者可以任意设定测序深度(sequencing depth,即基因序列总个数),而基因测序深度是影响Hi-C数据质量,分析效果强度的主要因素。此外,使用者可以通过调节模拟基因序列的错配(mismatches)、空位(gaps)、嵌合序列(chimeric reads)的比率来控制模拟数据与真实数据之间的相似度。

文章中,作者通过与Sim3C以及降采样(downsampling)的对比,展示出FreeHi-C对真实Hi-C数据结构各个层面(A/B compartment,TADs等)的精准模拟。同时,对diffHic【5】,multiHiCcompare【6】,FIND【7】,Selfish【8】四种差异信号检测(differential chromatin interaction detection)方法的比较分析中,FreeHi-C的推断结论一致,且不受测序深度的限制。最后,作者提出可以通过数据扩增(data augmentation),将模拟Hi-C数据加入差异信号检验中,以克服生物学重复(biological replicate)数量过少的问题。FreeHi-C模拟数据的加入极大地提高了检测的精度和强度,同时控制错误发现率在理想范围内(下图)。

原文链接:
https://doi.org/10.1038/s41592-019-0624-3
参考文献
1. Dekker, J., Rippe, K., Dekker, M., Kleckner, N.:Capturing chromosome conformation. Science 295(5558)(2002)1306–11
2. Lieberman-Aiden,E.,VanBerkum,N.L.,Williams,L.,Imakaev,M.,Ragoczy,T.,Telling,A., Amit, I., Lajoie, B.R., Sabo, P.J., Dorschner, M.O., et al.: Comprehensive map -ping of long-range interactions revealsfolding principles of the human genome. Science 326(5950) (2009)289–293
3. Rao,S.S.P.,Huntley,M.H.,Durand,N.C.,Stamenova,E.K.,Bochkov,I.D.,Robinson,J.T., Sanborn,A.L.,Machol,I.,Omer,A.D.,Lander,E.S.,Aiden,E.L.:A3Dmapofthehuman genome at kilobase resolutionreveals principles of chromatin looping. Cell 159(7)(2014) 1665–1680
4. DeMaere, M.Z., Darling, A.E.: Sim3c: simulation ofhi-c and meta3c proximity ligation sequencingtechnologies. GigaScience 7(2)(2017)gix103
5. Lun, A.T., Smyth,G.K.: diffHic: a Bioconductor package to detect differential genomicinteractions in Hi-C data. BMC Bioinformatics 16(1) (2015)258
6. Stansfield, J.C., Cresswell, K.G., Dozmorov, M.G.: multiHiCcompare: jointnormalization and comparative analysis of complex hi-c experiments.Bioinformatics(2019)
7. Djekidel,M.N.,Chen,Y., Zhang,M.Q.:Find:differentialchromatininteractionsdetectionusing a spatial poisson process. Genome Research 28(3) (2018)412–422
8. Ardakany, A.R., Ay, F., Lonardi, S.: Selfish: discovery ofdifferential chromatin interactions via a self-similarity measure.Bioinformatics (2019)i145–i153