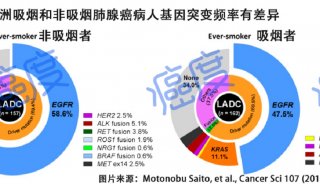
基因突变致蛋白质合成异常分析(七)
2.抗凝血因子缺乏症
(1)遗传性抗凝血酶Ⅲ缺乏症:抗凝血酶Ⅲ(antithrombin Ⅲ,ATⅢ)对凝血酶Xa有抑制作用,肝素能加速其对凝血酶的抑制。其次,ATⅢ还有抑制Ⅸ、Ⅺ及Ⅻ的功能。
遗传性抗凝血酶Ⅲ缺乏症(hereditary antithrombin Ⅲ deficiency)的临床表现为容易发生血栓形成(主要部位为髂静脉)及肺体塞。
本病为常染色体显性遗传。发病率在不同种族有显著差异。欧美白种人中可高1:2000-5000。我国也已有个例报告。现知,ATⅢ基因定位1q23,基因长16kb,由7个外显子组成,编码432个氨基酸,至少已发现20种以上的突变类型,表现出不同的功能缺陷。
(2)遗传性蛋白C系统异常:遗传性蛋白C系统由蛋白C(PC)、蛋白S(PS)、血栓调节蛋白(thrombomdorlin,TM)及活性蛋白C抑制物(APC1)组成,它们之间的关系见图4-19。活化的蛋白C有抑制Va、Ⅻa及促进纤维蛋白溶解的作用。蛋白C系统缺乏症有3种:
1)蛋白C缺乏症:主要症状是静脉血栓形成,常见于深部静脉。本病发生率估计为1:16000,呈常染色体显性(或不完全显性)遗传,纯合子严重,表现为出血性皮肤坏死、弥漫性血管内凝血(DIC)和血栓。杂合子多数在青壮年发病。蛋白C基因定位2号染色体,基因长12kb,由8个外显子组成,已鉴定出若干种缺失型及点突变病例,也有涉及mRAN加工缺陷者。
2)蛋白S缺乏症:蛋白S的作用是促进活化蛋白C(APC)结合于磷脂,加速APC灭活Va因子。本病亦属常染色体显性(或不完全显性)遗传。基因定位在3号染色体,其总长度为45kb。上海也已报告2例PS缺乏症。
3)先天性活化蛋白C抑制缺乏症(congenital deficiency of activated protein Cinhibitor)。
其它还有遗传性纤溶系统异常,包括①先天性异常纤溶酶原血症;②先天性纤溶酶原激活物释放异常;③遗传性纤溶酶原激活抑制物增多症;④血块异常所致先天性纤溶减弱等,均表现出高凝状态的各种症状。
五、受体蛋白病
受体是存在于细胞膜上、胞质中或核内的一类具有特殊功能的蛋白质。现已证明具有调节生理功能和特异性受体达30种以上。其中包括多肽激素受体、固醇类激素受体以及神经递质、前列腺素、免疫性因子、脂蛋白等受体。由于受体的本制是蛋白质,不言而喻,基因突变也可导致受体蛋白质和量的改变。由于受体蛋白遗传性缺陷引起的一类疾病,称为受体病(receptor diseae)。
受体病有获得性与遗传性之分。遗传性受体病研究得较多的有下列4种:
1.家族性高胆固醇血症 家族性高胆固醇血症(familial hypercholesterolemia,FH)的临床表现是:出生时即存在高胆固醇血症,增高的胆固醇主要为低密度脂蛋白胆固醇()LDL-C)和β极低密度脂蛋白(β-LDLC),黄色瘤(xanthoma)即增高的胆固醇在组织广泛沉积。其最严重的后果是早年发生动脉粥样硬化。纯合子在5-30岁即表现心绞痛和心肌梗塞症状,可骤死。杂合子发生冠心病稍迟且发生率较低。
本病为常染色体显性遗传病。在人群中,杂合子发生率为1:500,杂合子临床表现较轻,故属不完全显性遗传,外显率高(90%-100%)。
本病的主要病因是LDL受体(LDLR)的遗传性缺乏。血浆中的LDL和β-VLDL与LDL受体结合后,内吞入细胞,在溶酶体中LDL与LDLR分离,LDLR回到细胞表面重新利用,LDL则在胆固醇酯酶作用下,释出胆固醇供细胞利用。在这一过程中,细胞表面有功能的LDLR数量决定了血浆中LDL及β-VLDL的浓度。现知LDLR的编码基因位于19p1.3-p13.3,总长度45kb,由18个外显子组成。已发现FH患者有数十种突变,包括缺失型突变(主要的)、错义突变、无义突变、移码突变及整码突变。表现为LDLR功能异常有4种类型:①细胞膜上完全无LDLR;②LDLR明显减少;③LDLR无功能不能与LDL结合;④LDLR与LDL结合后不能内吞,可能是由于合成受体前后有加工缺陷。
2.睾丸女性化综合征 睾丸女性化综合征(testicular ferminization syndrome)又称雄性素全不敏感综合征(complete androgen insensitivity syndrome,CAIS),为最常见男性假两性畸形。患者核型为46,XY,有睾丸,能分泌雄性素,但缺乏雄性素受体,雄性素不能发生效应(图4-20)。