-
IF=27! Hi-C,发表高分Paper的开挂神器,你需要了解一下!
2018-12-03 11:31:15
Jesse R. Dixon, Jie Xu ,et al. Integrative detection and analysis of structural variation in cancer genomes. Nature Genetics, 2018, 50: 1388–1398. IF 27.125
主要技术:Hi-C、集成光学映射(Irys)、全基因组测序(WGS)
研究背景
结构变异(SVs),包括倒置、删除、复制阳离子和易位,是大多数癌症基因组的标志。复发性SVs的发现及其对基因组织和表达的分子效应促进我们对肿瘤发生的认识。许多致癌基因已被确认为复发易位的产物,并为药物治疗特别是造血恶性肿瘤提供了成功的靶点。尽管它很重要,但在癌症基因组中鉴定SVs仍然具有挑战性。作者利用高通量染色体构象捕获(Hi-C)、集成光学映射和全基因组测序,系统地检测正常或癌症样本中的SVs,意图探究癌基因组中,SVs对突变驱动因素的影响。
研究内容及结果
1. 检测肿瘤基因组SV的方法
为了评估对SVs不同检测方法的能力,作者选取了8个癌细胞系和1个典型正常对照(GM12878)(见表1),对它们的WGS、光学测图和Hi-C数据进行比较(图1a),发现三种方法均检测到Caki2细胞中染色体2和3的易位(图1b),通过观察同一区域DNA复制时间谱的显著变化,也证实了这种易位。同时观察到,与正常细胞相比,癌症基因组显示出更多的重组事件,如图1c所示环状基因组结构剖面。
表1肿瘤和正常细胞系的高置信度的SVs数
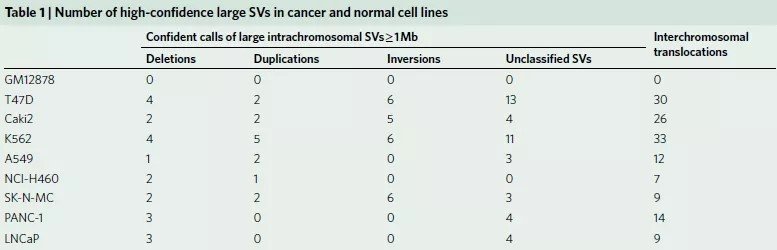
图1 肿瘤基因组SV检测的总体策略

2. 利用Hi-C数据检测大规模重排
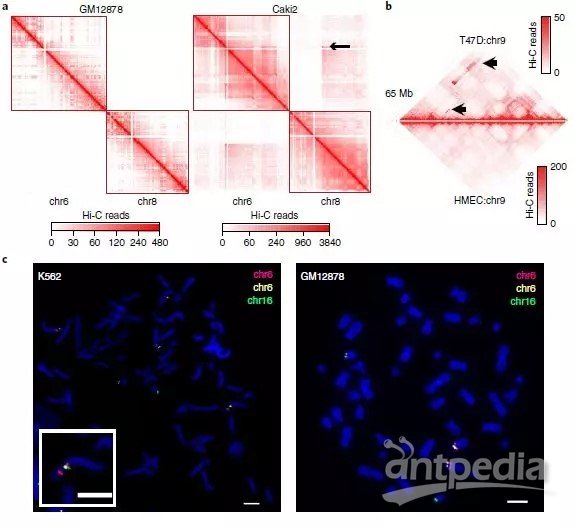
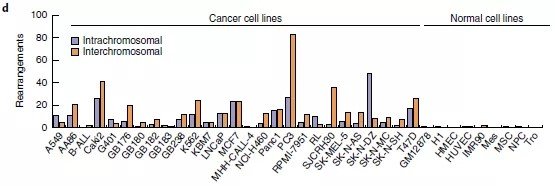
在Hi-C实验中,正常细胞染色体间相互作用非常罕见(图2a左)。然而,这这种情况在癌细胞中却相反。例如Caki2癌症在细胞中,观察到了强烈的染色体间相互作用(图2a右),这可能是由于6号染色体和8号染色体的融合。但是关于癌细胞的染色体相互作用增加的信号是由于重组还是三维基因组组织的变异导致的还不清楚,因此针对这一问题,作者首先为“正常”的三维基因组组织特征建立了概率模型,包括位点、TADs、A/B compartments之间的基因组距离,发现小染色体和次端粒区域之间相互作用的增加。并且在重排的情况下,两个重排区域的基因是融合的,因其改变了位点之间的线性距离,从而也导致了与局部预期交互频率的偏差(图2a、b)。其次,作者利用Hi-C数据进行全基因组SVs检测,这一检测属于一种新型算法。该算法具体体现为:作者首先用一个特征良好的慢性粒细胞白血病细胞系(K562)来评估,并将结果与已发表的核型进行比较。在19个Hi-C预测的重排中,11个可以确认,其余8个是新的。由于这8个均在两个独立实验室进行的,它们不太可能是克隆进化的产物。随后,作者进行了FISH实验来验证新的预测易位。使用Hi-C数据预测的19个易位中有18个通过FISH或以前的核型验证,结果表明,新算法能够识别具有高特异性的大规模结构变异。
最后,将Hi-C分析扩展到27个癌细胞系和9个核型正常细胞系(图2d),发现在癌细胞中报告了25次重排,在正常细胞中几乎没有发生这种情况,染色体间和染色体内重排的比率约为2:1(所有细胞系中为424比274)。因此,新算法似乎可以识别大部分的大SVs,只有4.3%的无法识别。
图2 利用Hi-C数据检测大规模重排
3. 不同方法检测SVs的比较
通过光学映射和WGS在每个癌细胞株中鉴定了数千个遗传物质的增加或损耗,光学映射检测到的缺失比WGS更少但范围更大。在T47D细胞中,WGS检测到2943个缺失,中位大小为552 bp,而Irys检测到1128个缺失,中位大小为1 335 bp(图3a,b)。其中85% WGS检测到的缺失被Irys遗漏,且其中78%的中位大小小于1kb。由于其分辨率受两个刻痕点之间的最小距离的限制,这些特征很可能被光学映射所忽略。Irys预测的缺失中有3%与多个较小的WGS缺失重叠,在这些情况下,这些WGS缺失的总和大小接近Irys检测到的缺失,但在Irys检测到的缺失中,有15%没有被WGS捕获。
作者测试了Irys检测到的一部分缺失,其中87.5%的缺失(16个缺失中的14个)通过了PCR验证。光学映射可以识别WGS reads没有被映射的重复区域内的缺失(图3c),以及在断点周围可映射性较低的区域。同时还发现WGS、Irys和Hi-C可以检测到不同染色体间大规模重排,类似基因组的非模板化添加碱基或外源DNA序列,如病毒的碱基,它可能来自第三条染色体因其太短而无法识别。如图3d所示,光学映射到局部结构,WGS用来确定断点,WGS通过定位断点和Hi-C数据来验证同一等位基因上几个相邻重排。总之,采用互补技术的综合方法对于更全面地了解癌症基因组的结构变化至关重要。
图3 不同方法检测SVs的比较

4. SVs对增强子的影响
Nature Genetics丨三维基因组学研究进展
2018-05-04 14:13:14
4月26日,武汉金开瑞生物工程有限公司技术顾问、华中农业大学教授, 曹罡课题组与华中农业大学李国亮教授课题组在国际著名期刊Nature Genetics上联合发表题为“Digestion-ligation-only Hi-C is an efficient andcost-effective method for chromosome conformation capture”的研究论文。该论文主要介绍了一种新的染色体构象捕获技术Hi-C,为解析基因组三维结构提供了一种新型、高效、经济的研究方法。 基因组三维空间结构与功能的研究简称三维基因组学(Three-Dimensional Genomics,3D Genomics),是指在考虑基因组序列、基因结构及其调控元件的同时,对基因组序列在细胞核内的三维空间结构,及其在基因转录、调控、复制和修复等生物过程中功能的研究。染色体是由DNA与组蛋白共同组成,从染色体的一级结构(绳珠模型)到四级超螺旋折叠结构,DNA分子一共被压缩了8400倍左右,正是这些折叠和压缩,导致基因在细胞中的分布复杂而又有序,只有了解清楚染色体区域(A/B compartments、TADs、Loops),才可以将基因组上原本分散的远距离调控元件与其具体调控区域更好的关联起来,对理解基因的转录调控、增强子与启动子的相互作用、疾病易感位点、DNA损伤修复、基因组结构变异和表观遗传有着重要的意义。科学家们对染色体三维结构的研究很早就开始了,但是相关的研究一直不够深入,同时缺乏微观的证据。直到2003年Job Dekker及其合作者提出了染色质构象捕获技术(Chromatin Conformation Capture,3C),用于测定特定的点到点之间的染色质交互作用。随后,科学家们扩展了3C技术,开发了4C技术(Circularized Chromatin Conformation Capture),用于测定一点到多点之间的染色质交互作用。Dostie等人接着开发了5C技术(Carbon-Copy Chromatin Conformation Capture),用于测定多点到多点之间的染色质交互作用。为了能捕获全基因组范围的染色质相互作用,Job Dekker研究组又开发出了现在大家所熟知的Hi-C技术。Hi-C技术是三维基因组学的主要研究方法,但存在实验成本高、数据噪音大、实验过程繁琐等缺点。为解决这些问题,曹罡教授及其合作者提供了一种新的DLO Hi-C染色体构象捕获技术(digestion-ligation-only Hi-C , DLO Hi-C),设计了巧妙的酶切位点,采用同时酶切酶连的方式,将DNA接头连接在染色体内切酶切口末端上,然后进行邻近酶连,最后再用MmeI内切酶酶切消化,回收固定大小互作DNA片段,从而大大地降低了测序背景数据噪音,获取的测序数据质量高于传统Hi-C。DLO Hi-C技术还可以用于染色体易位位点筛选。
基因组三维空间结构与功能的研究简称三维基因组学(Three-Dimensional Genomics,3D Genomics),是指在考虑基因组序列、基因结构及其调控元件的同时,对基因组序列在细胞核内的三维空间结构,及其在基因转录、调控、复制和修复等生物过程中功能的研究。染色体是由DNA与组蛋白共同组成,从染色体的一级结构(绳珠模型)到四级超螺旋折叠结构,DNA分子一共被压缩了8400倍左右,正是这些折叠和压缩,导致基因在细胞中的分布复杂而又有序,只有了解清楚染色体区域(A/B compartments、TADs、Loops),才可以将基因组上原本分散的远距离调控元件与其具体调控区域更好的关联起来,对理解基因的转录调控、增强子与启动子的相互作用、疾病易感位点、DNA损伤修复、基因组结构变异和表观遗传有着重要的意义。科学家们对染色体三维结构的研究很早就开始了,但是相关的研究一直不够深入,同时缺乏微观的证据。直到2003年Job Dekker及其合作者提出了染色质构象捕获技术(Chromatin Conformation Capture,3C),用于测定特定的点到点之间的染色质交互作用。随后,科学家们扩展了3C技术,开发了4C技术(Circularized Chromatin Conformation Capture),用于测定一点到多点之间的染色质交互作用。Dostie等人接着开发了5C技术(Carbon-Copy Chromatin Conformation Capture),用于测定多点到多点之间的染色质交互作用。为了能捕获全基因组范围的染色质相互作用,Job Dekker研究组又开发出了现在大家所熟知的Hi-C技术。Hi-C技术是三维基因组学的主要研究方法,但存在实验成本高、数据噪音大、实验过程繁琐等缺点。为解决这些问题,曹罡教授及其合作者提供了一种新的DLO Hi-C染色体构象捕获技术(digestion-ligation-only Hi-C , DLO Hi-C),设计了巧妙的酶切位点,采用同时酶切酶连的方式,将DNA接头连接在染色体内切酶切口末端上,然后进行邻近酶连,最后再用MmeI内切酶酶切消化,回收固定大小互作DNA片段,从而大大地降低了测序背景数据噪音,获取的测序数据质量高于传统Hi-C。DLO Hi-C技术还可以用于染色体易位位点筛选。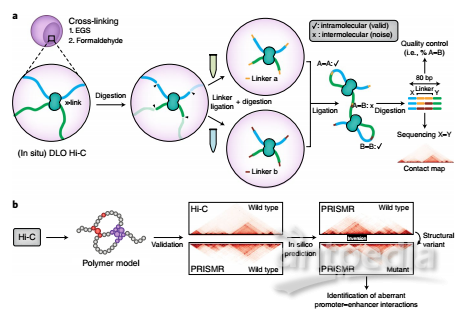 DLO Hi-C技术使全基因组染色体构象捕获实验的成本大大的降低,同时简化了实验步骤,使得实验成功率显著提高,对辅助基因组组装、解析基因组远程调控元件的功能、理解疾病易感位点以及检测染色体结构变异有着重要的意义。Nature Genetics同期发表专门评论性文章,对曹罡教授及其合作者的研究工作给予了较高评价“Lin et al. introduce an elegant dual-linkerstrategy that allows for noise filtering and early quality control.”
DLO Hi-C技术使全基因组染色体构象捕获实验的成本大大的降低,同时简化了实验步骤,使得实验成功率显著提高,对辅助基因组组装、解析基因组远程调控元件的功能、理解疾病易感位点以及检测染色体结构变异有着重要的意义。Nature Genetics同期发表专门评论性文章,对曹罡教授及其合作者的研究工作给予了较高评价“Lin et al. introduce an elegant dual-linkerstrategy that allows for noise filtering and early quality control.”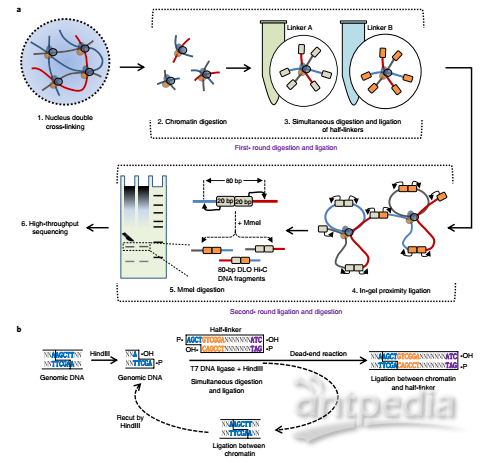 随后还将详细解读该篇论文,从实验设计、流程、生信分析及可视化方面进行详细说明,干货满满,敬情期待哦!文章链接:https://www.nature.com/articles/s41588-018-0111-2
随后还将详细解读该篇论文,从实验设计、流程、生信分析及可视化方面进行详细说明,干货满满,敬情期待哦!文章链接:https://www.nature.com/articles/s41588-018-0111-2

标题搜索
我的存档
数据统计
- 访问量: 0
- 日志数: 20
- 建立时间: 2017-11-09
- 更新时间: 2019-06-27
