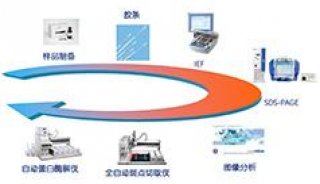
蛋白质技术——双向电泳
实验概要
蛋白质组研究的发展以双向电泳技术作为核心. 双向电泳由O’Farrell’s于1975年首次建立并成功地分离约1000个E.coli蛋白,并表明蛋白质谱不是稳定的,而是随环境而变化. 双向电泳原理简明,第一向进行等电聚焦,蛋白质沿pH梯度分离,至各自的等电点;随后,再沿垂直的方向进行分子量的分离. 目前,随着技术的飞速发展,已能分离出10000个斑点(spot). 当双向电泳斑点的全面分析成为现实的时候,蛋白质组的分析变得可行。
样品制备(sample prepareation)和溶解同样事关2-DE的成效,目标是尽可能扩大其溶解度和解聚,以提高分辨率. 用化学法和机械裂解法破碎以尽可能溶解和解聚蛋白,两者联合有协同作用。对IEF(isoelectric focusing)样品的预处理涉及溶解、变性和还原来完全破坏蛋白间的相互作用,并除去如核酸等非蛋白物质. 理想的状态是人们应一步完成蛋白的完全处理. 而离液剂2 mol/L硫脲和表面活性剂4% CHAPS的混合液促使疏水蛋白从IPG(immobilized pH gradients)胶上的转换. 三丁基膦(Tributyl phosphine,TBP )取代β-巯基乙醇或DTT完全溶解链间或链内的二硫键,增强了蛋白的溶解度,并导致转至第二向的增加。两者通过不同的方法来增加蛋白的溶解度,作为互补试剂会更有效。在保持样品的完整性的前提下,可利用超离和核酸内切酶去除核酸(DNA)。除此之外,机械力被用来对蛋白分子解聚,如超声破碎等. 另外,添加PMSF等蛋白酶抑制剂,可保持蛋白完整性. 由于商品化的IPG胶条是干燥脱水的,可在其水化的过程中加样,覆盖整个IPG胶,避免在样品杯中的沉淀所致的样品丢失。此外,低丰度蛋白(low abundance protein)在细胞内可能具有重要的调节功能,代表蛋白质组研究的“冰山之尖”,故分离低丰度蛋白是一种挑战。亚细胞分级和蛋白质预分级、提高加样量(已达到1~15 mg级的标准)、应用敏感性检测,可以提高其敏感性. 如一种多肽免疫2-DE印迹(MI-2DE)是利用几种单克隆抗体技术来分析和检测。提高组蛋白和核糖体蛋白等碱性蛋白(basic proteins)的分离是另一难点。由于碱性pH范围内凝胶基质的不稳定及逆向电渗流(EOF)的产生,对PI(等电点)超过10的碱性蛋白,通过产生0~10% 的山梨醇梯度和16%的异丙醇可减少之。亦可用双甲基丙烯酰胺来增加基质的稳定性。
2-DE面临的挑战是高分辨率和重复性。高分辨率确保蛋白最大程度的分离,高重复性允许进行凝胶间配比(match)。对2-DE而言,有3种方法分离蛋白:1)ISO-DALT(isoelectric focus)以O’Farrell’s技术为基础. 第一向应用载体两性电解质(carrier ampholyte, CA),在管胶内建立pH梯度。随着聚焦时间的延长,pH梯度不稳,易产生阴极漂移。2) NEPHGE(non-equilibrium pH gradient electrophoresis)用于分离碱性蛋白(pH>7.0). 如果聚焦达到平衡状态,碱性蛋白会离开凝胶基质而丢失. 因此,在等电区域的迁移须在平衡状态之前完成,但很难控制. 3)IPG-DALT发展于80年代早期. 由于固相pH梯度(Immobilized pH gradient, IPG)的出现解决了pH梯度不稳的问题. IPG通过immobiline共价偶联于丙烯酰胺产生固定的pH梯度,克服了IEF的缺点,从而达到高度的重复性. 目前可以精确制作线性、渐进性和S型曲线,范围或宽或窄的pH梯度. 新的酸性pH 3~5或碱性pH 6~11的IPG凝胶梯度联合商品化的pH 4~7的梯度可对蛋白质形成蛋白质组重叠群(proteomic contigs)从而有效分离。
分离后的斑点检测(spot detection)亦很重要. 所采用的检测策略和分离后所采用的方法的相互作用是很重要的. 此外,还需考虑反应的线性、饱和阈/动态范围、敏感性、对细胞蛋白群的全体定量分析的适应性、可行性. 目前,没有一种蛋白染色覆盖广泛的浓度和PI及分离后分析技术. 银染已成为一种检测2-DE的流行方法,可检测少到2~5ng的蛋白,因此较考马斯亮蓝R-250敏感. 多数糖蛋白不能被考马斯亮蓝染色,一些有机染料不适于PVDF膜. 放射性标记不依赖其代谢的活性,并仅适于对合成的蛋白质检测. 另有一种改良的2-DE(差异凝胶电泳),即应用两种不同的染料荧光标记两个样品,使在同一凝胶上电泳后的凝胶图象为两个,避免了几种2-DE的比较,可在纳克级进行检测。
较早期相比,2-DE有两个主要的进步:首先,极高的重复性使有机体的参考图谱,可通过Internet获得,来比较不同组织类型、不同状态的基因表达;其次,高加样量使得2-DE成为一项真正的制备型技术。
主要试剂
Acetic acid、 EtOH、 CBB、 琼脂、 丙烯酰胺SDS 等
样品提取液A:10%三氯乙酸,0.07%β-疏基乙醇的丙酮溶液
样品提取液B:0.07%β-疏基乙醇的丙酮溶液
样品提取液C:9.5mol/L尿素,2%NP-40,2%Ampholine (1.6%pH5~8,0.4% pH 3.5~10),5%β-疏基乙醇
丙烯酰胺液D:28.38%丙烯酰胺,1.62%甲叉双丙烯酰胺
凝胶平衡液E:10%甘油,0.1%β-疏基乙醇,4%SDS,0.1mol/LTris-HCl pH 6.8)
丙烯酰胺液F:29.1%丙烯酰胺,0.9%甲叉双丙烯酰胺
分离胶缓冲液G:1.5 mol/LTris-HCl (pH 8.8),0.4% SDS
浓缩胶缓冲液H:0.5mol/LTris-HCl (pH6.8),0.4% SDS
电极缓冲液I:1% SDS,3.22% Tris,14.1%甘氨酸
考马斯亮兰染色液J:50%乙醇,5%冰乙酸,0.2%考马斯亮兰R-250
银染显色液K:6%Na2CO3,含0.5mL37%甲醛/L,4mgNa2S2O3·5H2O/L
实验步骤
第一向(等电点聚焦,IEF)
1)凝胶条的准备
1.以帽凝胶端(2个校准环:4mm和10mm)朝下,将4根玻璃管插入两个凝胶灌注装置的每个托架中,这样玻璃管恰好站在两个分隔室之一的“充填船”上。从玻璃管顶端到玻璃管底端拉入PP线(小心,线不易移动),否则以后将不能用它们将胶溶液拉上来。
2.溶解分离胶溶液和过硫酸铵溶液。必须监控凝胶溶液的温度,因为高温下(大于25℃)聚合作用太快。
3.分离胶溶液除气4分钟,在桌子边缘轻弹玻璃管壁,避免产生气泡。
4.融化帽凝胶溶液,除气,步骤同3。
5.用Gilson移液管加35μl APS到1365μl分离胶溶液中,溶液应小心摇晃,避免氧气进入胶内。
6.将分离胶溶液加到两个凝胶灌注装置的每一个充填船里(每4个管700μl)。所有的玻璃管末端都必须浸没于凝胶溶液中。
7.小心将PP线抽出,将凝胶溶液拉上至23mm的标记。
8.换充填船分隔室。
9.使用一个Gilson移液管,加10μl 0.8% APS到390μl帽凝胶溶液中,轻轻振荡使之混匀。
10.在两个凝胶灌注装置的每个充填船中的空余分隔室内加入200μl帽凝胶溶液;凝胶溶液应到达17mm的标记。
11.移去充填船,允许凝胶溶液到达13mm的标记。经过这一步,有空气进入管子底端和4mm标记处。
12.聚合30分钟后,将PP线抽出,去除凝胶表面未聚合的溶液。在毛细血管口部滴加一大滴去离子水,这样在凝胶的上边形成一个潮湿的室,在凝胶和水滴之间形成一个气泡。用此方法形成一个潮湿的室,随后用Parafilm膜封上帽凝胶一侧。凝胶在室温过夜,可以使之进一步聚合。制备好的管子如果保存于室温,可以在第二天使用,或最迟在制备好第五天使用。
2)加样
1.将乙二胺加入阴极溶液,除气5min,下槽装阴极溶液。
2.融解Sephadex溶液,加入108mg尿素和10μl两性电解混合液到100mg Sephadex胶中,在涡旋器上充分振荡10~15分钟。
3.从凝胶灌注装置移走凝胶管,把它们插入聚焦槽的阳极部分,仍应看到13mm标记。帽凝胶一端朝上。
4.将阴极溶液加入管子的阴极一边,不要有气泡(事先已除去水)。
5.将聚焦槽的阳极部分安放到聚焦槽的底部,管子必须浸入阴极溶液。
6.除去水,并将加样侧干燥。使用一个伸长的巴斯德移液管或凝胶装填移液管头将水吸去,然后用滤纸条将凝胶表面干燥。
7.融解准备好的样品和覆盖溶液。
8.用凝胶装填移液管头,迅速在胶上覆盖大约2mm厚的Sephadex。
9.用一个凝胶装填移液管头,装入样品(1~15μl),加到Sephadex表面。加样时,尖端必须接触Sephdex表层,样品应小心加入,避免气泡。
10.使用凝胶装填头,轻轻在样品上加入5μl覆盖液并分层,不要使覆盖液和样品相混合。然后在毛细血管中加入阳性溶液,不要引入气泡。
11.所剩的阳极溶液加入上槽,用缓冲液覆盖所有的管子。
3)IEF电泳
电泳应分段升高以按下表获得Vh(电压×小时)乘积为1841:
电压(V) | 时间(分钟) | 电压(V) | 时间(分钟) |
100 | 75 | 600 | 75 |
200 | 75 | 800 | 10 |
400 | 75 | 1000 | 5 |
凝胶平衡
1.融解平衡溶液(在IEF结束前1小时)。使用之前,在20ml平衡溶液中加入0.2g DTT。
2.IEF结束后,移去管子中的阳性和阴性溶液。
3.用87%的甘油溶液覆盖帽(cap)凝胶一侧。
4.加5ml平衡溶液到Petri盘。
5.用PP线挤出凝胶:PP线从帽凝胶一侧插入,管子加样一侧浸入去离子水,小心地向下挤出凝胶。与此同时可看到水中的覆盖液的溶解。凝胶被拉出5min后,用水迅速清洗挤压端。然后将管子保持在盛有平衡溶液的Petri-盘子上,整个凝胶被PP线拉到盘子里。每个盘子里可平衡两块胶。
6.室温振荡平衡凝胶10分钟整。
7.IEF之后,如果凝胶并不立刻使用,应倒出平衡缓冲液,并在Petri盘中-70℃保存。-20℃保存导致分辨率降低;液氮保存将破坏凝胶。
第二向(SDS-PAGE)
1)SDS-PAGE胶的制备
1.加热琼脂糖溶液至70℃使之液化。
2.冷却琼脂糖溶液至40℃,保持液态。
3.喷洒70%乙醇清洗玻璃板和隔板,并用Kimwipes刷干净。
4.融解凝胶溶液和APS。
5.将玻璃板夹入夹紧装置,该夹紧装置放在灌装支架的前面狭槽中,这样旋钮对着支架,而“尖角”向上。充分旋转旋钮以使厚的有机玻璃正好对齐后部边缘。这时大玻璃板可以放在有机玻璃板的前面,隔板沿着左边和右边的边缘。插入小板,用拇指轻压玻璃板和隔板以确使玻璃板位于底部。用钳子夹紧做好的“三明治”,后部边缘出现Newton环说明旋钮已经非常紧了。不要将旋钮旋得过于紧,否则玻璃板会被夹碎。将三明治从支架抽出,用拇指检查玻璃板和隔板是否与底部齐平。
6.在小玻璃板底部起172.5px处做标记,做为加样得辅助(凝胶长度为165px)。
7.将凝胶“三明治”装置夹到灌装支架上,旋钮向内,“尖角”向上。在有机玻璃板上加压以使凝胶三明治插入小突起下面。
8.将18ml凝胶溶液和1.2ml APS相混合,不要有气泡。溶液必须混匀,不要引进氧气。
9.含有凝胶溶液得容器放在加样槽的后面玻璃板上,这样凝胶溶液可以沿着加样槽从玻璃板流下而没有气泡,分离胶被加到标记处。
10.迅速用去离子水覆盖凝胶溶液,以利于聚合并使表面平坦。在凝胶一端用一巴斯德移液管防止搅动凝胶溶液,并使水在凝胶表面均匀地分层。60分钟后,凝胶可以用于SDS-PAGE电泳。
2)加样
1.用滤纸条去除凝胶表面的去离子水。
2.将凝胶“三明治”放到电极装置上(旋钮冲外,尖角向上):首先将凝胶“三明治”放到电极装置的上面部分,向电极装置挤压它使下面一部分固定。
3.在平板凝胶表面加IEF凝胶:从平衡缓冲液中取出凝胶条,纵向放到厚有机玻璃板的边缘,尽量使凝胶条与加样槽的边缘靠近。使用小刮勺,将凝胶条轻轻向下推,使之小心的滑入加样槽。凝胶条必须位于SDS-PAGE的表面,没有气泡。
4.使用Pasteur移液管,将加样槽加满40℃的琼脂糖溶液(没有气泡),2分钟后,琼脂糖溶液应凝固。
5.在电极装置装入凝胶“三明治”以后,将Bio-Rad槽注入620ml电极溶液,加溶液的时候避免泡沫和气泡的形成。内槽中电极缓冲液面应到达电极装置中红塑料点的位置。
3)SDS-PAGE电泳
1.在将槽封闭之前,需要确定有机玻璃表面是否干燥,以避免形成“渐进”电流,凝胶表面必须覆盖电极缓冲液。
2.电压必须如下表所示分段升高,电流和功率要调到电源的最大值。电流值如表所述并在每一部内逐渐减小。在给定的时间间隔初始和结束的数值如下表所示:
电压 | 时间 | 一个槽中的电流 (2块胶) | 两个平行槽中的电流 (4块胶) |
35V | 5分钟 | 22~21mA | 44~42mA |
55V | 10分钟 | 33~32mA | 66~64mA |
100V | 15分钟 | 59~48mA | 118~96mA |
150V | 60分钟 | 72~50mA | 144~100mA |
3.SDS-PAGE结束之后,倒出电极缓冲液,从电极装置中取出凝胶“三明治”。轻微地弯曲隔板,将小玻璃板拉开以取出凝胶。凝胶可以转移到固定溶液中。
4.固定之后,可以采用几种染色方法。
注意事项
1.上样量的问题,我觉得我跑的这张2D图,上样量还需要调整,可以适当降低,我的ipg胶条是13厘米的上样量在50ng-80ng之间,上样量不合适,丰度低的将会被丰度高的所遮盖,这是最讨厌的问题。
2、跑一向的时候大家都差不多,根据公司的说明就可以做了。跑二向的时候我现在一般做恒压,以前做过恒流,没感觉有什么差异,还想请教各位!横流和恒压到底哪个更好一些!
3、ipg胶条ph的选择,我做的是植物的花药,一般情况下酸性端点多一点,碱性端较少,我现在选用的是ph3-10的,我觉得很不合适,(不过这个胶条是免费的)因此好多点没能很好的分开,如果胶条的ph小一点,胶条(我希望能达到450px)可是我们这里做不了,效果应该比这个好很多的!
4、从这块胶上,我觉得致命点就是背景深,将影响我下一步的分析。所以银染还需要改进!
5、针对不同的蛋白质,分离胶的浓度也是可以调节的,我比较懒,就作过10%,12.5%的,不过觉得都不适合。