激肽释放酶的研究进展
高血压
高血压可由收缩血管物质过多或舒张血管物质缺乏引起。BK能诱导血管内皮产生舒张因子,如一氧化氮(NO)和PGI2等,从而引起扩张血管,降低外周血管阻力及调节肾脏组织对钠盐的排泄,参与机体血压的调节。BK具有强大的利尿钠效应,可使肾脏血流量增多,肾小管周围毛细血管压增高,抑制肾小管再吸收,并通过刺激入球小动脉压力感受器及致密斑而产生利尿钠作用。另一方面,BK可抑制远端肾小管对钠和水重吸收及抑制抗利尿激素的作用,从而促进水钠排泄。在原发性高血压和肾性高血压患者中,血管对BK的降压反应明显增强,从而提示高血压状态下缺乏内源性激肽。KKS中很多成分不足可导致BK的产生减少,从而引起高血压。1934年德国科学家Elliot等首次发现动脉压升高患者尿KLK排泄减少。30多年后Margolius和Sharma等肯定了这一发现并观察到部分原发性高血压患者或自发性高血压大鼠(SHR)尿中KLK的水平都显著降低,而流行病学研究也发现,尿KLK水平与原发性高血压患者的血压呈反比。此外,一项针对心血管危险因素的遗传因子的研究提示肾或尿KLK基因显性表达可降低高血压发生的危险[9]。黑人较白人尿中KLK水平低,黑人高血压的发病率也较白人高。对非洲裔美国黑人的研究发现伴随着肾脏钾排泄的减少,尿KLK排泄在高血压前期就出现显著降低[9]。然而钾及醛固酮分泌水平恢复到与白种人(高加索人)相似的水平并不能使黑人的尿KLK排泄正常。这些发现提示非洲裔美国黑人KLK生物合成水平较低是由于KLK基因本身因素引起。许多动物模型也同样能发现尿KLK排泄减少,包括SHR及Dah1盐敏感性大鼠(DDS)。Sharma等[10]观察到过度表达肾脏组织KLK的转基因小鼠血压偏低,而给予组织KLK阻滞剂抑肽酶后可使血压恢复。激肽可降低低盐饮食的SHR的血压,而给予激肽B2受体拮抗剂后血压明显升高。激肽酶Ⅱ(ACE)抑制剂通常被应用于临床及实验性高血压的治疗,它的抗高血压作用与抑制激肽的生物降解及阻断血管紧张素Ⅱ在肾脏的形成,从而使体内激肽水平增高有关,因为同时应用激肽抗体,其降压作用便明显削弱。激肽原缺乏大鼠(brown Norway-Katholiek)因不能产生激肽而在接受血管紧张素Ⅱ、高盐饮食或脱氧皮质酮和钠盐刺激后比正常大鼠更早出现高血压[11]。从以上客观事实,可以得出这样的结论,除原发性醛固酮增多症这种因盐皮质类固醇分泌过多的高血压外,其余的高血压都可能与肾脏KKS功能低下有关。BK的降压作用通常是经过B2受体介导的。Sharma等[12]研究发现B2受体拮抗剂B5630能够阻断BK的降压作用,此外还能抑制血管紧张素转换酶抑制剂(ACEI)卡托普利的降压作用,由此可推断ACEI的降压作用除减少BK的分解而增加血液中浓度外,还能阻止激肽B2受体失敏,促使受体功能上调[13]。较早的研究显示,口服猪胰腺KLK可明显降低高血压患者的血压[14],其缺点为降压作用短暂,须反复给药。给实验动物静脉注射提纯的组织型KLK可引起快速而短暂的降压效应[15],BK受体抑制剂艾替班特(HOE140)能阻断此反应。已有多个应用组织KLK基因治疗高血压、逆转左心室肥厚(LVH)、减轻肾功能损害等各种高血压模型的研究展示转基因治疗降压效果持久,且对心血管及肾脏疾病具有良好的保护作用。
心肌缺血
激肽与内皮细胞B2受体内结合,释放NO及PGI2,发挥扩张血管和抗增殖效应,保存心肌高能磷酸物,增加对糖原的摄取和利用以对抗血管紧张素Ⅱ的作用,从而发挥维持心血管内环境稳定的作用。有证据表明KKS功能失调在心力衰竭的发病机制中发挥重要作用。Whalley等[16]报道心力衰竭心脏中微血管局部激肽生成减少,NO浓度下降。此外,在起搏诱导的狗的充血性心力衰竭模型中可观察到在使用艾替班特选择性阻断B2受体后冠状动脉血流及心肌收缩力下降,左心室舒张末压升高[17] 。因此可以认为心血管KKS活性降低促进心力衰竭的发展。另一方面,缺血预适应是一种心肌保护现象,是指心肌经1~4次短时间(2~10 min)缺血对随后的长时间缺血性损伤产生耐受性,细胞的损伤明显减轻。其机制至今尚未完全阐明,已有的研究证实,缺血可触发内源性自我保护,释放一系列内源性活性物质,BK便是其中之一。BK在缺血早期就由心肌组织释放,局部及全身性给予外源性BK可明显增加冠状动脉及毛细血管血流,改善心肌代谢[18]。Scholkens[19]研究证实在狗的冠状动脉内注入BK能显著降低缺血诱导的严重心律失常。BK冠状动脉灌注可提高结扎冠状动脉的SHR及WKY大鼠的存活时间,且该作用可被艾替班特抵消,提示BK对缺血预适应的心脏保护作用是由B2受体介导,通过激活信号转导途径产生NO和PGI2实现的[20]。对大鼠、狗及人的多个研究显示激肽在心肌及全身缺氧缺血状态下持续释放,特别是急性心肌梗死后,这一过程提示激肽在心肌梗死时发挥保护作用。药理学和遗传学研究显示KLK和激肽受体在缺血后被诱导活化是机体的自发性反应,以增加患部血液灌注,促进康复。因此急性心肌梗死后心肌BK量增加被认为是减少或限制梗死面积、增加心脏电稳定性、抑制再灌注心律失常的有益反应。随着分子生物学和基因定位的研究,越来越多的证据表明KKS在心血管病理生理学中的重要性,也为我们发展以KKS为基础治疗心血管疾病提供了可能。
凝血功能
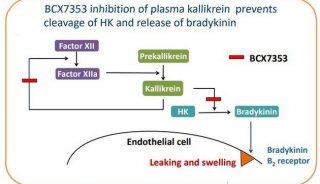
研究认为KKS部分作用与RAS系统相反[5]。在生理条件下,内皮细胞及其基质提供丝氨酸蛋白酶PRCP,激活激肽释放酶原(PK)转变为KLK。随后激肽释放酶激活FX Ⅱ[21],在与BK的前体激肽原结合后,无活性的PK转化为KLK。接下来,PRCP介导的向KLK的转化使激肽原转变为BK。BK 与内皮细胞B2受体结合,刺激细胞内Ca2+动员,引起NO和前列腺素的释放。通过同一受体,缓激肽还增加内皮储存池组织型纤溶酶原激活剂(t?PA)释放[22],发挥抗凝血作用。AngII刺激内皮细胞中纤溶酶原激活物抑制1(PAI?1)mRNA的表达,以剂量依赖方式升高血浆PAI?1水平[23],发挥促进血栓形成的作用,PRCP可降解Ang Ⅱ从而使PAI?1水平降低。血浆KLK可促进单链尿激酶和纤溶酶原的活化,发挥抗血栓形成作用。激肽原及降解产物也具有抗凝血酶活性。BK降解产物缓激肽(1?5)能通过与蛋白酶活化受体1和4上的凝血酶裂解部位结合,抑制凝血酶诱导的血小板聚集[24]。FXⅡ,PK和HMWK是参与内源性凝血的蛋白质,Merlo等[25]采用对照的方法对200名心肌梗死患者血中FXⅡ,FXⅠ,PK和HK的含量进行测定,发现FXⅠ,HMWK和PK水平显著升高,表明它们对心肌梗死的发生可能起一定的作用。
LVH
LVH被认为是高血压患者的独立危险因素。BK能够对抗主动脉结扎引起高血压大鼠LVH的发展,这种抗心肌肥厚的效应能被B2受体拮抗剂艾替班特殊性及NO合酶抑制剂L?NNA抵消,说明BK是通过降低NO释放来发挥抑制LVH的作用,证实在SHR大鼠LVH的发病机制中心血管KKS的缺乏占有重要作用,而心血管组织KLK及激肽原的活性降低是导致心脏BK减少的原因,因此心脏KKS组分的不足可能引起高血压和LVH心肌功能障碍。在用ACEI雷米普利进行降压及逆转LVH的观察中发现,大剂量雷米普利[1 mg/(kg·d)]共6周的治疗可降低血压,抑制LVH的发展,而给予小剂量雷米普利[10 μg/(kg·d)]6周后对血压及血浆中ACE活性没有影响,但阻止了主动脉结扎后引起的LVH。2种剂量雷米普利抗心肌肥厚的作用及大剂量雷米普利的降压作用均可被B2受体拮抗剂艾替特及NO合酶抑制抵消。该研究说明KKS和RAS在阻止或延缓心室肥厚这一靶器官损害的重要作用,更支持了KKS是心血管保护因子的观点。
-
项目成果