







Fast and reliable mini-prep RNA extraction from Neurospora crassa
We have developed a method for isolating high quality total RNA from N. crassa mycelia that reliably yields large quantities. It is possible to extract more than 50 minipreps at once.
Procedures of RNA extraction published so far follow roughly three different approaches, where phenol/chloroform, guanidinium salts, and/or LiCl is used. However, these protocols have some disadvantages. They are either time consuming, or yield low amounts of RNA, or are not powerful enough for cultures containing high levels of nucleases. Extraction from starved cultures or plate mycelia with the LiCl method (Chambers and Russo, Fungal Genet. Newsl. 1987. 33:22-24) gave completely degraded RNA as judged by gel electrophoresis. In trying to improve the method we modified the protocol used for Chlamydomonas by Gromoff et al. (Mol. Cell. Biol. 1989. 9:3911-3918) who combined the advantages of phenol extraction and LiCl precipitation. The procedure is suitable for rapidly grown mycelia, starved mycelia, mycelia from plates (4 days old) and single colonies of N. crassa grown on a sorbose plate. With this protocol, from 20 to 300 mg (dry weight) of mycelium, grown in liquid media or on plates, yielded between 200 µg and 3 mg of RNA depending on the growth conditions and strains. From a single colony we could isolate up to 5 µg total RNA.
Mycelium from a liquid or surface grown culture was harvested, washed, dried thoroughly with filter paper and frozen in liquid nitrogen. Single colonies including agar were stamped out using an inverted Pasteur pipette and frozen.
The procedure of extraction given below is described for Eppendorf tubes but it is also possible to scale up the volumes. All manipulations were performed at room temperature if not stated otherwise.
- Pulverize the mycelium in a mortar with liquid nitrogen
- Transfer the powder into a mixture of 0.75 ml lysis buffer (0.6 M NaCl, 10 mM EDTA, 100 mM Tris HCl, pH 8.0, 4 SDS) and 0.75 ml phenol (saturated with 0.1 M Tris HCl, pH 8.0) in a 2 ml Eppendorf tube. The tube can be filled with powdered mycelium
- Shake for 15-20 min (Eppendorf Rotationsmischer 3300)
- Centrifuge for 10 min, 10,000 rpm
- Transfer the upper phase into an equal volume of phenol (saturated with 0.1 M Tris HCl, pH 8.0) and vortex
- Centrifuge for 10 min, 10,000 rpm
- Add 0.75 volumes of 8 M LiCl to the upper phase
- Store overnight at 4°C
- Vortex briefly and centrifuge for 10 min, 10,000 rpm
- Resuspend the pellet, which is not always visible, in 0.3 ml double distilled water, mix with 0.03 ml 3 M Na-acetate (pH 5.2) and 0.75 ml ethanol
- Store at -20°C for 2 h or at -70°C for 30 min
- Centrifuge for 10 min, 10,000 rpm
- Discard the supernatant and wash the precipitate with 70 ethanol
- Dry the RNA pellet and redissolve it in DEPC treated (diethyl polycarbonate) water
- Store the RNA solution at -70°C
Purity of the RNA preparations was assayed by spectrophotometric measurements. The A260/A230 and A260/A280 ratios were 2 or more, indicating the absence of any protein or polysaccharide contamination. Usually, traces of DNA present in RNA preparations can be seen in the slots of ethidium bromide stained formaldehyde gels. Based on this criterion, RNA samples prepared by the described method were free from contaminating DNA, while clear nondegraded ribosomal bands were seen in all 200 extractions we made (see Figure 1). Northern blot analysis by hybridization with several 32P-labelled probes (Figure 2) and in vitro translation experiments (Figure 3) have shown clear signals indicating the presence of intact mRNA.
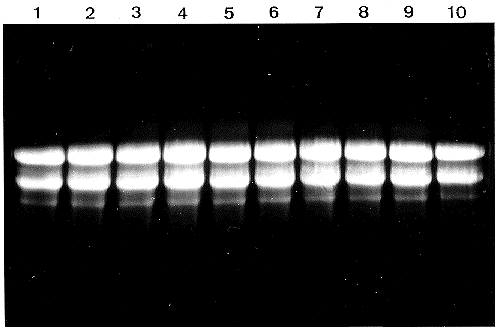
Figure 1. Total RNAs (10 礸 per lane) from 10 different preparations (lanes 1-10) were electrophoresed on a 1.2 agarose gel containing 2 formaldehyde. Visualization was done by staining with ethidium bromide and irradiation with UV. The major bands correspond to 28S and 18S rRNA, faint bands to 23S, 16S and 5S rRNA.
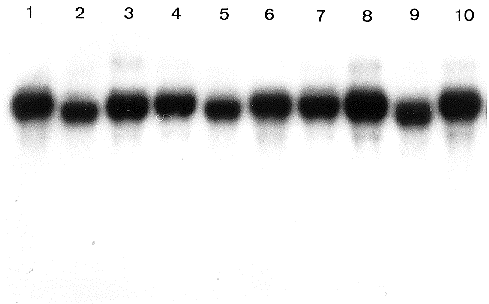
Figure 2. Autoradiogram after Northern blotting and hybridization to 32P-labelled cDNA insert of arbitrarily chosen clone N6 (T. Sommer et al. 1989. Nucl. Acids Research 17:5713-5723). Northern analysis was done according to R.A. Kroczek and E. Siebert 1990. Analyt. Biochem. 184:90-95.
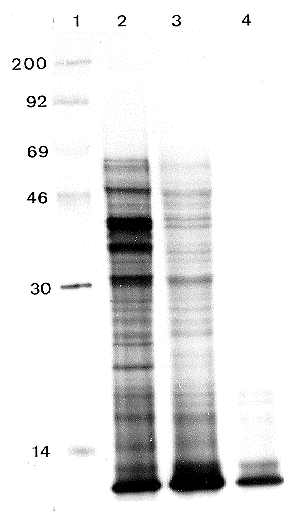
Figure 3. SDS-PAGE (12.5) of 35S-methionine-labelled in vitro translation products. Lane 1: 14C-labelled marker proteins, molecular wieghts are indicated on the left (Mr x 10(-3). Lanes 2 and 3: RNA prepared from mycelia grown in rich or ammonium depleted medium respectively. RNA probes correspond to lanes 1 and 2 in Fig. 1 and 2. Lane 4: control translation without RNA.
Acknowledgements: We thank Beate Otto for in vitro translation experiments and Niketan Pandit for critically reading the manuscript.
