巨噬细胞在内皮细胞调控肌肉再生新机制中的应用
内皮细胞(endothelial cell,EC)遍布血管,是氧气和营养物质输送、免疫细胞运输以及清除远组织废物的重要管道。目前认为内皮细胞(ECs)代谢是促进血管生成的重要因素,ECs不同的代谢途径,如糖酵解、脂肪酸氧化和谷氨酰胺代谢,在血管形成过程中发挥了不同的重要作用。研究发现,糖酵解过程中的一个关键限速酶——磷酸果糖激酶-2/果糖-2,6-二磷酸酶3(phosphofructokinase-2/fructose-2,6-bisphosphatase 3,PFKFB3)具有较强的激酶活性,若抑制其活性可以显著减少糖酵解,从而抑制病理性血管的生成。ECs还通过血管分泌信号传导促进组织稳态和再生,然而,EC是否参与代谢性血管内分泌串扰以控制局部缺血诱导的肌肉再生尚不清楚。
近期,苏黎世联邦理工学院的 Katrien De Bock团队(第一作者为张静)在Cell Metabolism杂志上发表了题为“Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization”的文章,揭示了内皮细胞通过分泌乳酸介导巨噬细胞极化来促进血管重生和肌肉再生,证明了在肌肉内存在第二种乳酸穿梭机制,即从内皮到巨噬细胞。

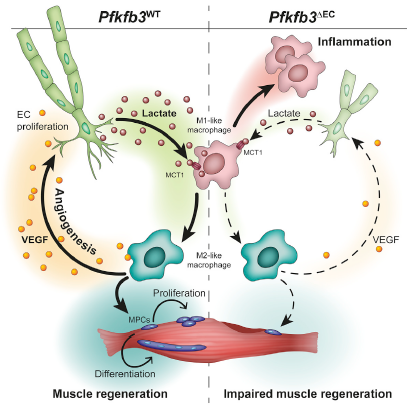
巨噬细胞在骨骼肌再生中起关键作用。研究人员发现,糖酵解调节因子pfkfb3在内皮细胞(EC)的特异性缺失可以缓解缺血性后肢受损的血管生成和肌肉再生能力。这是由于巨噬细胞促血管生成和促再生的M2型表型的能力降低引起的。为了剖析内皮细胞PFKFB3特异性敲除小鼠中M2型极化受损与缺血后肌肉修复受损的功能相关性,后肢缺血3天后,研究人员将未极化的骨髓来源巨噬细胞或M2型巨噬细胞注射到小鼠后肢,发现M2型巨噬细胞显著促进内皮细胞PFKFB3特异性敲除小鼠肌肉修复,但并未完全达到对照组水平。说明 内皮细胞PFKFB3敲除后对缺血后肌肉组织修复的影响是部分通过对M2型巨噬细胞极化调控发挥作用。
为了研究EC是否利用血管分泌机制来影响巨噬细胞极化,研究人员分离了野生型小鼠内皮细胞和PFKFB3敲除小鼠内皮细胞与BMDMs共培养,发现野生型小鼠内皮细胞显著促进BMDMs分化成M2型巨噬细胞。有趣的是,用mECs-CM(条件性培养基)进行的BMDM刺激不能完全概括经典的IL-4介导的M2极化。从mEC的CM中清除代谢物会减弱其诱导M2型极化的能力。通过对正常内皮细胞和PFKFB3敲除内皮细胞来源培养基的细胞因子(cytokine)和代谢产物分析发现内皮细胞敲除PFKFB3后,细胞因子无显著变化,而乳酸分泌显著下降。同时在补充生理浓度乳酸后,PFKFB3敲除内皮细胞来源培养基促进M2型巨噬细胞极化能力增强。值得注意的是, 乳酸控制巨噬细胞极化的能力需要条件培养基,这表明功能性极化需要其他细胞因子或代谢物的存在。
研究人员进一步收集了分化后巨噬细胞培养基,发现PFKFB3敲除内皮诱导的巨噬细胞培养基分泌更多的VEGF。通过促进M2型巨噬细胞极化(通过移植M2型巨噬细胞转移或增加乳酸水平)来恢复内皮细胞PFKFB3特异性敲除小鼠肌肉中的VEGF水平,可增加血管密度,尽管后者在施用乳酸后未能达到野生型小鼠的水平。这表明观察到的血管生成缺陷(至少在缺血性肌肉中)是PFKFB3对内皮迁移/增殖的直接抑制作用以及肌肉微环境对血管生成刺激减少的组合。
单羧酸盐转运蛋白1(MCT1)是乳酸的重要运载体,为了研究内皮细胞来源乳酸促进巨噬细胞极化的分子机制,研究人员用巨噬细胞MCT1特异性敲除小鼠(赛业生物构建)进行了更深一步的研究。发现在下肢缺血条件下,巨噬细胞MCT1敲除抑制M2型巨噬细胞的分化,同时降低了肌肉组织VEGF的分泌。病理学分析发现MCT1敲除抑制血管新生和肌肉组织再生。放射性标记实验发现巨噬细胞以MCT1作为乳酸运载体将内皮来源乳酸转运到细胞内,并作为底物参与到巨噬细胞氧化代谢。在低氧和营养不足的条件下为巨噬细胞发挥功能提供能量。
总之,该研究证明了内皮细胞利用其独特的代谢特征在缺血期间以依赖于乳酸穿梭至巨噬细胞的血管分泌方式调控肌肉再生。乳酸介导的巨噬细胞极化促进血管生成和肌肉再生。因此,EC特异性pfkfb3的丢失会降低肌肉乳酸水平并损伤缺血后肌肉的修复。代谢性血管内分泌信号提供了一种新型机制,通过该机制,EC可以促进组织稳态和再生。
原文检索:
Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization.
DOI: 10.1016/j.cmet.2020.05.004.