蛋白质组学实用分析技术一览
分析其实也属于技术的一部分,且在蛋白质组学研究中显得尤为重要,因为蛋白质组学研究提供的数据是生物学上最庞大的,而且蛋白质组比基因组具有更大的复杂性,因此蛋白质组信息学更有挑战性。今天小编先抛砖引玉地介绍几个常用的分析方法和软件。
蛋白质定性分析
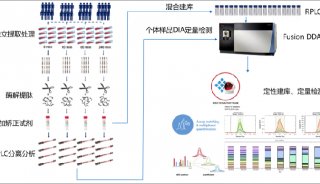
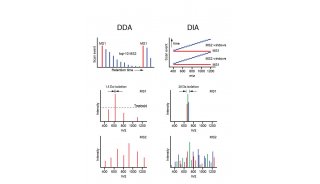
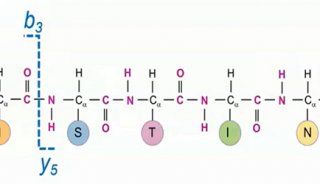
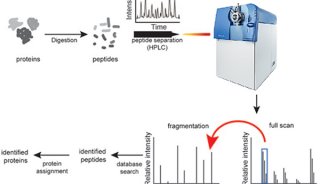
蛋白质定性分析通常是指利用质谱法进行蛋白质鉴定和序列分析。蛋白质定性分析分为top-down分析和bottom-up分析两种策略。目前,bottom-up分析策略被更广泛的应用于蛋白质定性分析工作。
根据使用仪器可分为:MALDI-TOF/TOF、LC-ESI-MS/MS
根据样本类型可分为:蛋白质全谱分析(即shotgun分析) 、蛋白质胶条/混合液分析、蛋白质胶点分析
蛋白质全谱分析
定义:蛋白质全谱分析也可称为质谱shotgun分析,是指组分分析,能够鉴定出尽可能多的肽和蛋白质分子。
原理:将溶液内蛋白质分子或SDS-PAGE条带的复杂混合物酶解成肽段混合物,通过液相色谱分离。串联质谱测试,最后用相应的数据库进行检索匹配,可同时鉴定成百上千种蛋白质。主要应用于某一生理状态下组织、细胞或者细胞器中所有表达蛋白质的鉴定。

蛋白质胶条/混合液鉴定
定义:利用LC-ESI-MS/MS蛋白鉴定技术对胶条样本(即SDS-PAGE样本)、IP、Co-IP、Pull-down等纯化溶液等中等复杂样本进行蛋白鉴定。
原理:利用电喷雾(ESI)技术将样品以离子化的方式从液滴表面蒸发,进入质量分析器。主要应用于蛋白质或多肽鉴定。
蛋白质胶点鉴定
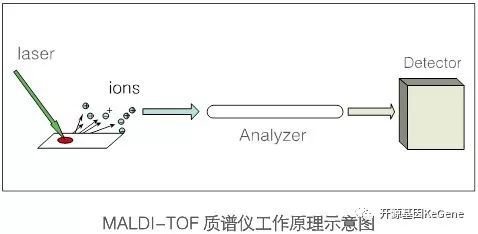
定义:利用MALDI-TOF/TOF蛋白鉴定技术对考/银染的2D或DIGE胶点样本或者较纯的蛋白样本进行蛋白鉴定。
原理: MALDI-TOF/TOF(Matrix – Assisted Laser Desorptiob/Ionization Time of Flight Mass Spectrometry,基质辅助激光解析电离飞行时间质谱)MALDI的原理是将样品均匀包埋在基质中,基质吸收激光提供的能量而蒸发,携带部分样品分子进入气相,并将一部分能量传递给样品分子使其离子化。TOF的原理是离子在电场作用下加速飞过飞行管道,根据到达检测器的飞行时间不同而被检测,即测定离子的质荷比(M/Z)与离子的飞行时间成正比,从而检测离子。 也主要应用于蛋白质或多肽鉴定。
蛋白质相对定量分析
相对定量的目的是测定目的蛋白在两个或多个样本中的表达量的相对比例,而不需要知道它们在每个样本中的表达量。例如,如果研究项目中包括处理过的和未经处理的对照样本,通常可以将未经处理的样本指定为基准,规定其目的蛋白浓度为100%,经处理的样本的定量结果除以对照样品的定量结果,就可以计算各个处理样本的蛋白含量相对于未处理样品的百分比。
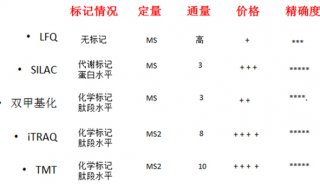
| 方法 | 原理 | 优势 | 应用 |
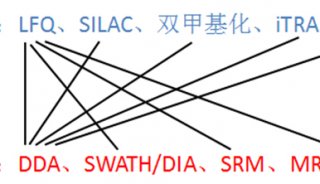
| iTRAQ | 采用4种或8种同位素的标签,通过特异性标记多肽的氨基基团,然后进行串联质谱分析。 | 可同时对2-8种不同条件样品进行蛋白质差异分析。适合广泛样品类型:胞浆蛋白、膜蛋白、核蛋白、胞外蛋白等。 | 疾病标志物筛选、作用机制研究、植物抗逆研究、药物作用靶点研究、殊功能蛋白质筛选。 |
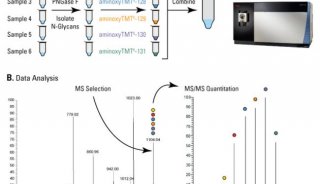
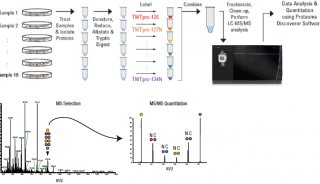
| TMT | 采用2种、6种或10种同位素的标签,通过特异性标记多肽的氨基基团,然后进行串联质谱分析。 | 可同时对2-10种不同条件样品进行蛋白质差异分析; 适合广泛样品类型:胞浆蛋白、膜蛋白、核蛋白、胞外蛋白、分泌蛋白等。 | 疾病标志物筛选、分子机制研究、植物抗逆研究、药物作用靶点研究、特殊功能蛋白质筛选。 |
| Label-free | 通过液质联用技术对蛋白质酶解肽段进行质谱分析,无需使用稳定同位素标签做内部标准,只需分析大规模鉴定蛋白质时所产生的质谱数据,比较不同样品中相应肽段的信号强度,从而对肽段对应的蛋白质进行相对定量。 | 蛋白质不需要进行标记,所需样品总量少,耗费低。 | 疾病标志物筛选、作用机制研究、物抗逆研究、药物作用靶点研究、殊功能蛋白质筛选 |
| SILAC | 分别用天然同位素(轻型)或稳定同位素(重型)标记的必需氨基酸取代细胞培养基中相应氨基酸,细胞经5-6代倍增周期后,稳定同位素标记的氨基酸完全掺入到细胞新合成的蛋白质中替代了原有氨基酸。不同标记细胞的裂解蛋白质按细胞数或蛋白量等比例混合,经分离、纯化后进行质谱鉴定。 | 细胞培养时掺入标记,减小实验误差,定量更准确。 | 疾病标志物筛选、作用机制研究、物作用靶点研究、特殊功能蛋白质筛选。 |
| DIGE荧光差异双向凝胶电泳 | 荧光差异双向凝胶电泳(DIGE)是一种在2D电泳之前标记蛋白质样品的方法,可以对样品间蛋白质丰度进行精确分析。 | 相对应传统的双向电泳技术,检测灵敏度更高,大大节约样品;采用内标对数据进行归一化处理,消除胶与胶之间差异造成的误差,准确度高。 | 疾病标志物筛选、作用机制研究、植物抗逆研究 、药物作用靶点研究、特殊功能蛋白质筛选。 |
蛋白质绝对定量分析
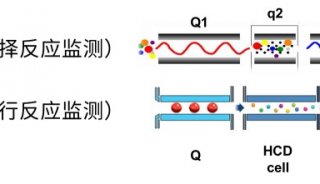
目前基于质谱的绝对定量蛋白质组学研究主要是指靶向蛋白质组学。靶向蛋白质组学分析,是指对目标蛋白质(或修饰肽段)进行定性和/或定量分析,或者用于验证大规模蛋白质组学的结果。基于质谱的靶向蛋白质组学分析方法,由于没有物种限制并具备多目标同时分析能力等优势,在相关研究领域已逐渐受到越来越多的关注和应用。其技术方法经历了从传统的SRM/MRM(选择性/多反应监视,selected / multiple reaction monitoring)到PRM(平行反应监视,parallel reaction monitoring)的发展历程。
绝对定量蛋白质组学应用方向: 验证定量蛋白质组学结果、验证翻译后修饰蛋白质组学结果、 目标蛋白质绝对/相对定量分析、诊断标志物靶向筛选、诊断标志物验证与绝对定量、验证基因表达产物。
PRM
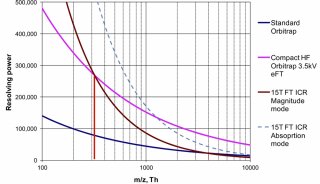
定义:PRM是一种基于高分辨、高精度质谱的离子监视技术,能够对目标蛋白质、目标肽段(如发生翻译后修饰的肽段)进行选择性检测,从而实现对目标蛋白质/肽段进行绝对定量。
原理:首先利用四级杆质量分析器的选择检测能力,在一级质谱中(Q1)选择性地检测目标肽段的母离子信息;随后在collision cell中对母离子进行碎裂;最后利用高分辨、高质量精度分析器在二级质谱中检测所选择的母离子窗口内的所有碎片的信息。这样即可对复杂样本中的目标蛋白质/肽段进行准确地特异性分析。
蛋白质翻译后修饰分析
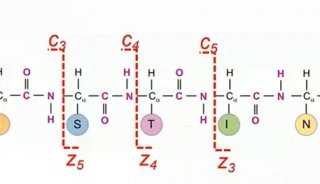
翻译后修饰(Post-translational modification, PTM)是指对翻译后的蛋白质进行共价加工的过程,通过在一个或多个氨基酸残基加上修饰基团,可以改变蛋白质的理化性质,进而影响蛋白质的空间构象和活性状态、亚细胞定位、折叠及其稳定性以及蛋白质-蛋白质相互作用。许多至关重要的生命进程不仅由蛋白质的相对丰度控制,更重要的是受到时空特异性和翻译后修饰的调控,揭示翻译后修饰的发生规律是解析蛋白质复杂多样的生物功能的一个重要前提。常见的翻译后修饰包括磷酸化、糖基化、乙酰化、泛素化等等。
修饰蛋白质组学:质谱是鉴定蛋白质翻译后修饰的重要方法,其原理是利用蛋白质发生修饰后的质量偏移来实现翻译后修饰位点的鉴定;同时,由于翻译后修饰的蛋白质在样本中含量低且动态范围广,检测前需要对发生修饰的蛋白质或肽段进行富集,然后再进行质谱鉴定。
修饰蛋白质组学技术方法及应用:
| 翻译后修饰 | 修饰位点 | 富集方法 | 生物学功能 |
| 磷酸化 | Ser,Thr,Tyr | 二氧化钛;酪氨酸磷酸化基序抗体 | 信号转导、细胞周期、生长发育及癌症机理等 |
| N-糖基化 | Asn | 凝集素 | 蛋白质折叠、更新以及免疫应答等 |
| 乙酰化 | Lys | 赖氨酸乙酰化基序抗体 | 基因表达调控、细胞凋亡、细胞代谢、蛋白质稳定性以及神经退行性病变等 |
| 泛素化 | Lys | 赖氨酸泛素化基序抗体(K-ε-GG) | 细胞周期、细胞凋亡、转录调控、DNA修复以及信号转导等 |
| 甲基化 | Lys,Arg | 赖氨酸/精氨酸mono/di/tri-甲基化基序抗体 | 染色质结构及转录调控等 |
| 琥珀酰化 | Lys | 赖氨酸琥珀酰化基序抗体 | 主要存在于线粒体和细菌中,与代谢过程密切相关 |
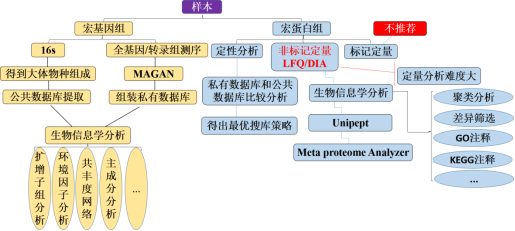
生物信息学分析
在这个大数据时代,生命科学的研究领域由还原论的研究方法逐步向系统水平过渡,作为高通量的各种组学研究手段在生命科学的研究中发挥着越来越重要的作用,并积累了海量的数据。如何从海量的生物数据中挖掘出有用的信息并指导生命科学的研究是整个生命研究领域的挑战。生物信息学在这个过程中起着越来越重要的作用。
生物信息学在蛋白质组学中的应用很广泛,通过双向凝胶电泳、生物质谱等方法和手段对蛋白质的种类、数量、结构和功能进行最后确定,也可对蛋白质的结构和功能进行预测。随着基因组研究的进一步拓展,蛋白质研究技术的不断革新和数据的不断积累,生物信息学工具也在日益完善,为蛋白质组学提供更丰富的资料。

蛋白质组学生物信息学主要内容:
蛋白功能注释:亚细胞结构定位、GO分类、KEGG通路途径、蛋白结构域、KOG/COG分类、FunCate2分类。
功能富集分析:使用费歇尔确切检验方法,描述特定属性蛋白群体(差异表达的蛋白、发生修饰的蛋白等)潜在调控的生理过程。分析包含:GO分类富集、KEGG通路富集、蛋白结构域富集、蛋白复合物富集。
功能富集聚类分析:使用分层聚类的方法,研究不同实验处理条件下对特定属性全蛋白群体调控生理过程的影响。
表达模式聚类分析:使用Mfuzz聚类方法对蛋白在不同处理条件(不同处理时间或药物浓度)下表达谱的聚类分析。直观的反应出处理时间或药物浓度与蛋白表 达水平的关系。对得到的不同表达模式分类可以做进一步的功能富集聚类分析,揭示药物潜在作用的蛋白与生理功能。
修饰位点motif分析:分析翻译后修饰位点附近氨基酸分布情况,预测潜在与该种修饰类型相关的保守序列区间。通过与蛋白结构域的联系,为研究该种修饰类型作用和调控机制研究提供参考依据。
蛋白质序列分析
Compute pI/Mw蛋白质序列的理论等电点、分子量的计算
CATH通过自动、手动方式进行蛋白质结构相关性的系统分类,“CATH”是以下四个单词的首字母的组合:
Class,类 —源于二级结构的最高级别的分类
Architecture,构造—独立于拓扑结构的二级结构排列描述
Topology,拓扑结构—参照已知的折叠及观测的结构进行拓扑分析
Homology,同源—结构间相同的拓扑布局、不同的二级结构(与功能相似性相关)描述
CRASP(Correlation Analysis of Sequences of Proteins)蛋白质序列的相关分析
FPAT检索分子序列数据库模式的简易方法
Hydropathy Plot(GIF image) 绘制亲疏水图
MOTIF蛋白质序列模式检索
RandSeq随机的蛋白质序列生长子
REPRO蛋白质重复片断识别服务器
SALSA蛋白质、核酸序列相似性检索
SignalP在不同的物种中预测信号肽及酶切位点
SIM 由用户定义的最佳两序列对比
TMpred蛋白质跨模区预测、定位,该方法基于统计学结果,通过权重矩阵打分进行预测分析
Coils对比分析具有双股卷曲结构的序列同源性打分,进而计算适于采用该结构的序列的几率
GuessProt选择SwissProt蛋白的等电点、分子量
MassSearch蛋白质消化后的聚集检索
Phospepsort磷酸化位点分类
SAPS蛋白质序列统计分析
Sleuth氨基酸构象及可及性分析
PEDANT蛋白质提炼
PROVE蛋白质原子体积计算
Naccess(ASA for proteins) 利用Lee & Richards方法描述分子表面性质的计算程序,能够计算蛋白质的表面可及性
蛋白质空间构象、同源模建
DOCK分子对接程序
Cn3D分子空间构象的三维模式展示程序
PROCHECK已知蛋白质结构的立体化学参数评价,通过分析残基间几何构象绘制图表。
Barton Group Home of the programs: AL, AMPS, AMAS, STAMP, ASSP and SCANPS.
WPDB 2.0蛋白质三维结构展示程序
CCP4蛋白质晶体解析程序
X-PLOR 适用于计算结构生物学的程序系统,通过经验能量函数及实验数据的限定,进行大分子空间构象的开发,该程序主要用于X-射线衍射数据及NMR核磁共振数据分析。
O 蛋白质结晶程序
MACAW (sequence alignment)多序列对比,功能块的定位、分析、编辑。
PDBtool- Macromolecular Structure Browsing and Verification - v1.0 Beta 蛋白质数据库的检索
Swiss-Model: Automated Protein Modelling Server 基于结构知识的蛋白质自动同源模建程序
VMD 1.0 (visual Molecular Dynamics)用于UNIX操作系统的、易于操作的分子图形学软件
XRayView适用于X-射线衍射晶体数据的交互式动态分析,涉及晶胞的构建,晶格的确定,系统消光,旋转摄影,空间群的确定及Laue群对称性等。
PDB (PROTEIN DATA BANK) 由实验决定的生物大分子三维结构的数据库
SCOP基于序列、结构的蛋白质结构分类数据库
NRL-3D源于PDB与PIR数据库的综合信息检索
CHEMICAL STRUCTURE DATABANK基于蛋白质化学结构分析
MMDB基于蛋白质结构数据库,由NCBI提供的分子模建数据库
RASTER3D光栅3D技术提供高质量的蛋白质及其他分子的光栅图像,利用有效的Z-buffer法则进行球状、三角、柱状展示,借助借助PDB提供的原子坐标进行飘带、球、球棍、键等的展示
vmd分子图像程序
Swiss-Model蛋白质自动同源模建
蛋白质二级结构预测、分析
DSC(Discrimination of Protein Secondary Structure Class) 蛋白质二级结构分类识别
DSSP 由Wolfgang Kabsch 与Chris Sander建立的标准的蛋白质二级结构分类,对蛋白质结构数据库PDB中的蛋白质进行归类
EpiMatrix/HIV 利用特定的抗原矩阵线性检索抗原表位
PredictProtein蛋白质二级结构预测
Garnier-Robson-Osguthorpe Secondary Structure Prediction 利用Garnier等建立的模型进行蛋白质二级结构预测
NAMDIllinois大学中的理论生物物理学会建立的提供高可信度的分子动力学模拟软件包
nnPredict蛋白质二级结构预测
PSSP蛋白质二级结构预测
Predator来自单序列或一套序列的蛋白质二级结构预测
SIMPA96 SECONDARY STRUCTURE PREDICTION蛋白质二级结构预测
SSPRED蛋白质二级结构预测
蛋白质相互作用分析
| 数据库名 | 说明 | 网址 | |
| BIND | 生物分子相互作用数据库 | http://bind.ca/ | |
| DIP | 蛋白质相互作用数据库 | http://dip.doe-mbi.ucla.edu/ | |
| IntAct | 蛋白质相互作用数据库 | http://www.ebi.ac.uk/intact/index.html | |
| InterDom | 结构域相互作用数据库 | http://interdom.lit.org.sg/ | |
| MINT | 生物分子相互作用数据库 | http://mint.bio.uniroma2.it/mint/ | |
| STRING | 蛋白质相互作用网络数据库 | http://string.embl.de/ | |
| HPRD | 人类蛋白质参考数据库 | http://www.hprd.org/ | |
| HPID | 人类蛋白质相互作用数据库 | http://wilab.inha.ac.kr/hpid/ | |
| MPPI | 脯乳动物相互作用数据库 | http://fantom21.gsc.riken.go.jp/PPI/ | |
| biogrid | 蛋白和遗传相互作用数据,主要来自于酵母、线虫、果蝇和人 | http://www.thebiogrid.org/ | |
| PDZbase | 包含PDZ结构域的蛋白质相互作用数据库 | http://icb.med.cornell.edu/services/pdz/start | |
| Reactome | 生物学通路的辅助知识库 | http://reactome.org/ |