Interleukin-6 Induced Acute Phenotypic Microenvironment Promote...(三)
Target analysis by Parallel reaction monitoring (PRM)
The targeted quantification and verification were carried out in 94 serum samples with 5~6 mice in each condition without pooling, across 8 time points. 23 proteins were selected with 1~4 peptides, resulting in 33 precursors in total (Additional File 1: Table S1). Glycopeptides were enriched as above and one of the angiotensin peptides was spiked in as the standard reference. PRM analysis was performed by Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific, San Jose, CA) with the same 2 h LC gradient setting as above. The MS acquisition mode was a combination of two scan events: a full scan and a time-scheduled scan. The full scan was taken at a resolution of 70000 at m/z 200 with a scan mass range of 200 to 1800 m/z, a target (AGC) of 1e6 and maximum injection fill time is 200 ms. The scheduled scan was employed at a resolution of 35000 at m/z 200, a target AGC of 5e5, and maximum injection fill time is 105 ms. The precursor ion of each target peptide was isolated with 2 Da window. The elution time window was set as +/- 5 min. Precursor ions were fragmented with normalized collision energy of 27%. The complete data acquisition took 15 days of mass spectrometer time.
Computational MS data analysis
Shotgun data dependent MS
MS raw files from DDA acquisition were analyzed by the Trans-Proteomic Pipeline software suite (TPP, Institute for Systems Biology, Seattle, WA) (version 4.7.0). Raw files were first converted to centroid mzXML and MS/MS spectra acquired were searched with comet (version 2.0) against a combined UniProtKB/Swiss-Prot and UniProtKB/VarSplic mouse database downloaded [29] with reversed sequences. Common contaminants [30] were included. Searching parameters were used as follows: precursor ion mass tolerance: ±10 ppm; fragment ion mass tolerance: 0.02 Da; semi-tryptic termini and two missing cleavages were allowed; fixed modification: carbamidomethylation (C); isobaric modification of lysine and peptide N-terminal residue; dynamic modification: oxidation (M) and deamidation (N). Searching results were further processed with PeptideProphet and ProteinProphet embedded in TPP. False discovery rate (FDR) for protein was set to 0.01. Isobaric labeling quantification was performed by LIBRA integrated in TPP and only unique N-glycopeptides with a PeptideProphet score ≥ 0.85, corresponding to an FDR <0.01 were considered for N-glycosylated protein quantification. The efficiency of glycopeptide capture was carried out and measured as the percentage of peptides with NXS/T sequence motif (X represents any amino acid residue except proline) in total peptides and corresponding glycosites were checked in Uniprot manually. The shotgun mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium [31] via the PRIDE partner repository with the dataset identifier PXD003136.
PRM MS analysis
MS raw data acquired from target proteomics analysis was searched with SEQUEST integrated in Proteome Discover (version 1.4, Thermo Fisher Scientific, San Jose, CA) against the same database and search parameters described above. Peptide probabilities were calculated by the Percolator algorithm in Proteome Discover and false discovery rate was set to 0.01. The generated .msf file was used to create a library in Skyline (version 3.1.0) [32] and the cut-off score was set as 0.90. The target precursors list was then uploaded to Skyline and the 6 most intense product ions matching the library were selected as transitions. Peak picking was manually checked and corrected according to the transitions, retention time, mass accuracy and MS/MS spectra. The area under curve (AUC) of each transition was extracted and exported to Excel for further data analysis.
Bioinformatics analysis and statistics
Shotgun MS data normalization
For shotgun MS data, the intensities of reporter ions were used as a proxy of protein abundances. Each protein’s abundance was divided by its channel’s global reference and the median value was obtained and centered to 1 to equalize the intensities between runs [33]. After normalization, we assessed the correlation of technical replicates for each sample, which well fit a line with an average R2 of 0.875 (Additional File 2: Table S2). In this case, protein’s abundances from two technical replicates were acquired and averaged to improve the quantification accuracy. For PRM MS data, each sample’s average base peak intensity was extracted from the full scan acquisition using RawMeat (version 2.1, VAST Scientific, www.vastscientific.com). The normalization factor for sample N was calculated as fN= the average base peak intensity of sample N/the median of average base peak intensities of all samples. The AUC of each transition from sample N was multiplied by this factor. After normalization, the AUC of each transition was summed to obtain AUCs at the peptide level. Relative protein’s abundance was defined as the intensity of a certain peptide.
Statistical analysis
Unsupervised hierarchical clustering analysis was handled in R package with default parameters for distance and similarity calculation. Ingenuity Pathway Analysis platform (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity) was used to analysis the enriched pathways and functional annotation. Other statistical analysis was performed in GraphPad prism 5 (version 5.01, San Diego, CA). Data were presented as the mean ± standard deviation (SD). A two-sample, two-side student t-test was applied to compare the difference between the mean values of two groups. Two-way ANOVA was used for multiple groups’ comparison with Bonferroni post-test.
Enzyme-linked immunosorbent assay (ELISA) for interleukin-6 (IL-6) assessment
IL-6 level in serum was measured by ELISA using commercial kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Serum samples were collected 2, 24, 48 and 264 h after treatment. Triple biological and two technical replicates were performed. Expression level was compared to that of mice untreated.
Neutralization of IL-6 in vivo
Mice injected with 4T1 cells were treated as described above. 100 μg of mouse anti-IL-6 Ab or control rat IgG Ab (Sanjian, Tianjin, China) was intraperitoneally injected to mice 3 and 24 h post cryo-thermal therapy. Mice were sacrificed 2 days post therapy and serum was collected.
Western blot analysis of acute phase proteins
Sera from different therapeutics (cryo-thermal /cryotherapy/hyperthermia/no treatment) and IL-6 neutralization were collected 2 days post therapy and subjected to 12% SDS-PAGE gel. Protein amount was evaluated with Ponceau S (Beyotime, Shanghai, China). Target proteins were immunoblotted with specific Abs as following: anti-APCS, anti-ORM1 (1:500, Absci, Baltimore, MD) and anti-HP (1:500, Proteintech, Rosemont, IL). Each protein was analyzed on three independent mice with three technical replicates. Blots were evaluated with Quantity One 1-D (Bio-Rad, Hercules, CA). Results are expressed as relative pixel intensity normalized with control group.
Quantitative reverse transcription–PCR
Real time PCR was applied to examine the level of IL-6 within primary tumors and IL-12, IFN-γ within spleens. 24 h or 3 days after therapy, primary tumors or spleens were harvested and homogenized with gentle MACS Dissociator (Miltenyi Biotec Technology, Bergisch Gladbach, Germany). RNA was extracted in Trizol (InvitrogenTM, Thermo Fisher Scientific, San Jose, CA) reagent and reverse transcribed to cDNA using Superscript III (Invitrogen, Thermo Fisher Scientific, San Jose, CA) according to the manufacturer’s instructions. β-actin was used as the control transcript. Primer sequences used were as follows: IL-6-FW5’-GACAAAGCCAGAGTCCTTCAGAGAGATACAG-3’; IL-6-RV5’-TTGGATGGTCTTGGTCCTTAGCCAC-3’; IL-12p40-FW5’-TGGTTTGCCATCGTTTTGCTG; IL-12p40-RV5’-ACAGGTGAGGTTCACTGTTTCT; IFN-γ-FW5’-CCATCCTTTTGCCAGTTCCTC; IFN-γ-RV5’-ATGAACGCTACACACTGCATC; β-actin-FW5’-CATGTACGTTGCTATCCAGGC-3’; β-actin-RV5’-CTCCTTAATGTCACGCACGAT-3’. Difference in expression of IL-6, IL-12 and IFN-γ compared to mice untreated was determined by calculating the fold change in expression (2−ddCT).
-
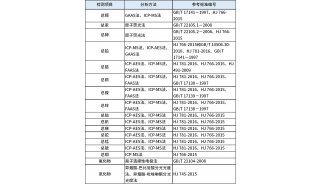
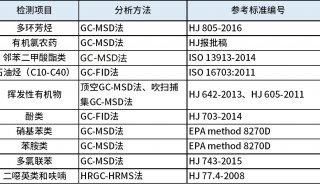
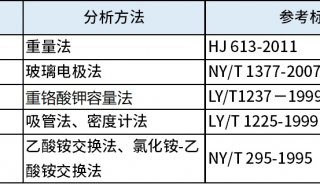
技术原理