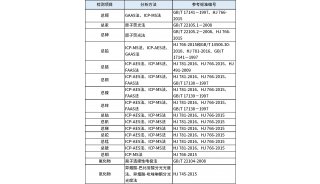
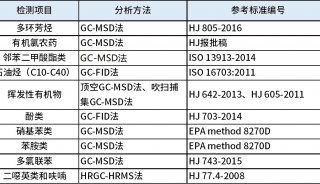
a pipeline for the identification of intact N-glycopeptides(三)
Figure 1. The overall workflow of pGlyco. First the sample is analyzed by HCD-MS/MS (NCE = 40%).
Then the product-dependent CID-MS/MS and data-dependent MS3 analyses are performed separately. pGlyco identifies glycopeptides by integrating the distinct information from these three complementary MS acquisitions based on the peptide backbone masses and the retention time. A solid line refers to data acquisition, and a dotted line refers to data interpretation.
pGlyco integrated spectral information from HCD-MS/MS, CID-MS/MS and MS3 to identify glycopeptides. We divided our MS data acquisition and analysis workflow into four steps, as illustrated in Fig. 1:
Step 1: HCD-MS/MS.
After full scan, precursors were firstly fragmented by HCD with normalized collision energy (NCE) at 40%. Diagnostic ions in an HCD-MS/MS spectrum were used to determine whether the precursor was a glycopeptide16,17, and then to trigger the succeeding acquisition of CID-MS/MS or DDA-MS3 spectra only for the true glycopeptides. We found that the peak 138.055 was always the highest peak in a HCD-MS/MS spectrum at 40% NCE, and it was specifically enough to select the true glycopeptide precursors by using 138.055 (see the section “Glyco-oxonium ions” in the Supporting Information). Moreover, in HCD-MS/MS at 40% NCE, the Y1 ion (the peptide backbone with a HexNAc attached) often coexisted with its corresponding cross-ring fragmentation on the HexNAc residue (i.e., the 0,2X0 ion)6,21, which was also considered as one of the trimannosyl core ions in pGlyco. There are 9 trimannosyl core ions used in pGlyco, which are Y0 (naked peptide), Y1 (peptide + HexNAc1), 0,2X0 (peptide + cross-ring fragment of HexNAc), Y2 (peptide + HexNAc2), Y3 (peptide + HexNAc2Hex1), Y4 (peptide + HexNAc2Hex2), Y5 (peptide + HexNAc2Hex3), Y_2 (peptide + HexNAc1dHex1) and Y_3 (peptide + HexNAc2dHex1), as defined in Figure S-1 in the Supporting Information. We named the 0,2X0 ion as Y1_ ion for simplicity.
Step 2: HCD-pd-CID-MS/MS.
HCD-pd-CID was performed to generate a CID-MS/MS spectrum with the same precursor of the HCD-MS/MS spectrum. For each HCD/CID-MS/MS spectrum pair, the total number of matched trimannosyl core ions was used as a feature to filter the candidate Y1 ion of each glycan in the glycan database, and the candidate peptide backbone mass could be deduced. Theoretical Y ions could be calculated as the deduced peptide backbone mass plus the masses of the reducing-terminal fragments of a glycan, and then they were matched and scored against the HCD/CID-MS/MS spectrum pair. A novel target-decoy method with a finite mixture model was used to estimate the false discovery rate (FDR) of the glycan identification.
Step 3: HCD-pd-MS3.
Taking the advantage of the novel instrument settings provided by the Orbitrap Fusion, MS3 spectra could be acquired for the three most intense peaks in the HCD-MS/MS spectrum within a certain mass range. And the Y1 ion may present as one of the most intense ions in HCD-MS/MS, which enables MS3 acquisition of Y1 ions to be performed in a data-dependent mode. MS3 spectra were identified by a protein identification search engine, pFind 2.8, and the conventional target-decoy approach for the peptide identification was employed to estimate the FDR. HCD-pd-MS3 was performed in another MS run to obtain more MS2 and MS3 spectra due to HCD-pd-MS3 need a much longer duty cycle.
Step 4: Data integration.
Information of MS/MS and MS3 spectral analysis from the previous three steps was assembled. Glycans identified by HCD- and CID-MS/MS spectrum pairs, and peptides identified by MS3 spectra were aligned based on the peptide backbone masses and the retention time. And then glycopeptides were identified with complete information of both glycans and peptide backbones.
Filtration of Y1 ions. The Y1 ion information was a bridge to connect the glycan identification and the peptide backbone identification for intact glycopeptides. For each glycan candidate (in the glycan database) of a given HCD/CID-MS/MS spectrum pair, the Y1 ion mass could be deduced by subtracting the glycan mass from the precursor mass of the spectrum pair (Y1 ion mass = precursor mass – glycan mass + HexNAc mass), and then the corresponding trimannosyl core ions could be calculated. At the beginning, pGlyco filtered out unreliable candidate Y1 ions by the following criteria: there must be at least three trimannosyl core ions matched in the HCD/ CID-MS/MS spectrum pair, or the (Y1, Y1_) ion pair with the same charge state was matched in the HCD-MS/MS spectrum. After filtration, a mass list of candidate Y1 ions were obtained for the spectrum pair, and the peptide backbone mass was deduced from the mass of each candidate Y1 ion.
Interpretation of glycans. For the glycan analysis, both HCD- and CID-MS/MS spectra were used. The scoring scheme for glycan identification of pGlyco was a revised version of the previously reported algorithm for the CID-MS/MS spectral analysis of glycopeptides6. With the peptide backbone mass deduced from the mass of each candidate Y1 ion, the masses of Y ions resulting from glycosidic bond cleavages were calculated by the mass of the peptide backbone plus the masses of the reducing-terminal fragments of each glycan structure, and then they were matched against the HCD/CID-MS/MS spectrum pair. The scoring scheme of pGlyco considered the matched peaks, their matching mass errors and the number of matched trimannosyl core ions, which was listed below:

The term inteni is the absolute intensity of a matched
peak. The term toli refers to the matching mass tolerance of fragment
ions, e.g. 20 ppm, and merri refers to the matching mass error ranging
from −to li to + to li. The score of each matched peak is weighted by a
quartic polynomial function, ,
which aims to penalize the larger mass errors with heavier penalties.
The term, ratioion, is the ratio of the number of matched ions to the
number of theoretical ions, and ratiocore is the ratio of the number of
matched trimannosyl core ions to the number of theoretical trimannosyl
core ions. By cross validation, the parameters α and β were fine-tuned
as 0.22 and 0.45 respectively. At last, the top-ranked glycan of each
spectrum pair was kept in the final results.
,
which aims to penalize the larger mass errors with heavier penalties.
The term, ratioion, is the ratio of the number of matched ions to the
number of theoretical ions, and ratiocore is the ratio of the number of
matched trimannosyl core ions to the number of theoretical trimannosyl
core ions. By cross validation, the parameters α and β were fine-tuned
as 0.22 and 0.45 respectively. At last, the top-ranked glycan of each
spectrum pair was kept in the final results.
-
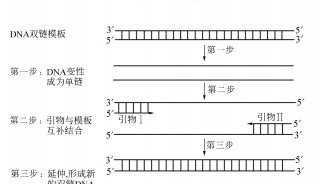
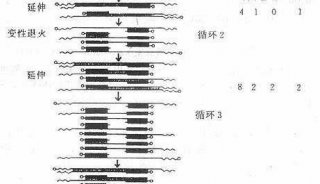
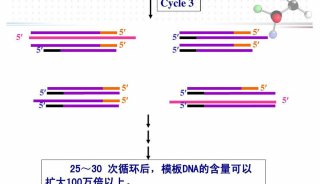
技术原理