基于Q-OT-qIT质谱系统的蛋白质组学快速鉴定(四)
A: 1 μg HeLa结果比较; B: 50 ng HeLa结果比较. 以每项最高值为100%
3 讨论
随着“一小时酵母蛋白质组”全覆盖的实现, 蛋白质组深度覆盖研究迎来新的时代[6]. 在复杂样本蛋白质组研究中, 传统方法需要进行二维分离、分级的方式简化样本、充分分离, 这样不仅操作费力, 也需消耗大量的时间. 而Q-OT-qIT质谱技术使得在60 min左右一维短梯度下实现复杂样本的深度鉴定成为现实.
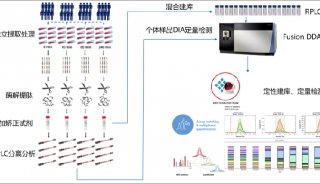
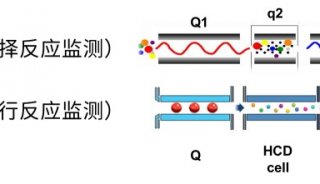
Q-OT-qIT的三合一结构实现了传统质谱难以实现的并列运行, 即一级、二级扫描同时进行, 离子注入、选择、碎裂、检测同时进行(图7). 动态扫描管理技术自动控制和实时优化仪器运行, 使3种质量分析器相互配合、同时工作, 尽可能减少扫描过程和循环间隙的等待时间. “减一逻辑”将二级扫描与上一次一级扫描相关联, 无需等待本次一级扫描确定母离子及电荷, 提高仪器运行速度和分析效率. 以top 10的数据依赖扫描(DDA)为例, 240k分辨率下, 当一级全扫描完成时, 所有二级扫描也同时完成, 一级、二级互不相关、独立运行(图7). 此外, top-speed模式在固定时间内尽可能多地激发二级扫描, 相比传统Top-N模式能更多地采集低丰度肽段, 也保证了一级扫描点数, 有利于同时定性定量.
实验结果表明, 1 μgHeLa样本在50 min有效梯度内采集到53209张谱图, 扫描速度为17.7 Hz, 其中包括120k分辨率的一级轨道阱扫描, 并列运行的工作模式提高了仪器整体运行速度. 在严格卡值(1% FDR)下, 鉴定到20860条非冗余肽和3865个非冗余蛋白, 而最新的文献报道需要至少2~3h才能达到相同结果[9]. 酵母样本虽未达到Hebert等人[6]报道的蛋白鉴定数量, 但上样量更低, 并且未进行重复, 比目前其他仪器的报道结果也有2倍以上的提高[8,9]. 在扫描速度方面, Q-OT-qIT虽然未达到Q-TOF类质谱的极限速度, 但谱图质量更高, 谱图解析率比Q-TOF类质谱高2~3倍[8].
1 μgHeLa样本在多次实验中的谱图解析率基本达到60%以上, 在加大离子注入时间和注入数量的情况下甚至达到83.1%(表3), 显示了良好的谱图质量. 全蛋白样本体系复杂, 短梯度下会有共流出肽段和基质杂质的干扰, 大量低丰度肽段的二级响应也很弱, 在这样的情况下, 谱图解析率达到80%以上, 显著高于目前的文献报道[8,9]. 此外, 样本量决定了蛋白鉴定数量, 传统蛋白质组学分析需要微克级的全蛋白, 而本实验尝试将样本量降低到纳克级, 即50 ng上样量, 鉴定结果已达到目前微克级的水平, 为低浓度样本和极低丰度蛋白分析提供可能.
Q-OT-qIT的三合一结构使扫描方式灵活多样. C-trap与离子阱之间的多极离子通道使离子可以正反向自由移动, 实现了CID/HCD/ETD3种碎裂模式在MSn任意一级使用, 并任意使用轨道阱或离子阱检测, 产生多种组合的扫描模式. 其中, CID碎裂相比HCD灵敏度略高, 但是速度略慢, 且有三分之一效应, 即低质量端歧视, 而HCD在低分子量段具有丰富的碎片信息; 轨道阱的超高分辨率与质量精度可以最大程度地排除干扰, 而离子阱速度更快, 并且可以和轨道阱同时运行, 提高效率. 可见, 由于机理的不同, 各种扫描模式的效果不同, 具有一定的互补性. Q-OT-qIT控制离子注入时间和注入数量的能力也是保证分析效率和灵敏度的重要因素. 注入时间越长、注入数量越多, 则灵敏度越高、谱图质量越高, 但扫描速度越慢; 相反, 离子注入数量越少, 速度越快, 但低丰度离子损失越大, 灵敏度下降. 可见, 实验目的和样本不同,适合的扫描模式和参数也不同, 因此, 实验进一步考察了扫描模式和离子注入参数对结果的影响.
结果表明, 不同碎裂模式和检测方法均具有良好的互补性. 轨道阱和离子阱之间有1/2的鉴定结果不重复, HCD和CID之间有2/3结果不重复, 不同的扫描方法结合可以达到更大的覆盖范围. 另一方面, 线性离子阱相比传统离子阱质量精度更高, 质量偏差稳定保持在0.2 Da以内, 达到中高分辨质谱水平, 因此, OT和IT扫描均具有较高的谱图解析率. 在离子控制方面, 通过提高最大注入时间和最大注入数量, 可以增加离子注入量, 提高低浓度样本和极低丰度蛋白的灵敏度; 而降低离子注入量, 能够加快常规样本的分析速度、增加鉴定数量. 通过几种模式和参数的考察、优化, 为蛋白质组快速分析和深度覆盖研究提供了更多选择.
4 结论
本文利用Q-OT-qIT三合一质谱, 对HeLa和酵母全蛋白进行了短梯度快速鉴定: (ⅰ) 50 min有效梯度, 从1 μgHeLa中鉴定到20860条非冗余肽和3865个非冗余蛋白, 时间相比目前的报道缩短了50%~60%; (ⅱ) 50 ng极低样本量, 鉴定到14100条非冗余肽和2877个非冗余蛋白, 谱图解析率超过50%; (ⅲ) 考察了HCD/CID碎裂模式和IT/OT检测方法的异同, 结果显示, 不同方法间互补性良好, 并且均有较高的谱图解析率; (ⅳ) 考察了离子最大注入时间和自动增益控制对结果的影响, 表明提高注入时间和注入数量可以显著提高低浓度样本的检测灵敏度. 综上所述, 基于Q-OT-qIT质谱, 本文实现了短时间内对复杂样本蛋白质组深度覆盖分析, “一小时蛋白质组”时代已经到来.

图7 Q-OT-qIT质谱结构与并列运行原理
轨道阱一级全扫描的同时, 四极杆依次进行若干母离子选择, 离子阱依次进行若干母离子的二级碎片扫描, 三者同时运行
致谢 部分图片素材由赛默飞世尔科技圣何塞工厂提供.
参考文献
1 Delahunty C M, Yates III J R. MudPIT: multidimensional protein identification technology. Biotechniques, 2007, 43: 563–569
2 Jones K A, Kim P D, Patel B B, et al. Immunodepletion plasma proteomics by triple TOF 5600 and orbitrap elite/LTQ-orbitrap Velos/Q exactive mass spectrometers. J Proteome Res, 2013, 12: 4351–4365
3 Mann M, Kelleher N L. Precision proteomics: the case for high resolution and high mass accuracy. Proc Natl Acad Sci USA, 2008, 105: 18132–18138
4 Geiger T, Wehner A, Schaab C, etal. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics, 2012, 11: M111.014050
5 Senko M W, Remes P M, Canterbury J D, et al. Novel parallelized quadrupole/linear ion trap/orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal Chem, 2013, 85: 11710–11714
6 Hebert A S, Richards A L, Bailey D J, et al. The one hour yeast proteome. Mol Cell Proteomics, 2013: M113.034769
7 Nagaraj N, Kulak N A, Cox J, et al. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top orbitrap. Mol Cell Proteomics, 2012, 11: M111.013722
8 Andrews G L, Simons B L, Young J B, et al. Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal Chem, 2011, 83: 5442–5446
9 Kelstrup C D, Young C, Lavallee, R, et al. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res, 2012, 11: 3487–3497
The One Hour Proteome: Q-OT-qIT Mass Spectrometry Based Rapid Proteome Identification
ZHANG Wei, GU PeiMing, JIANG Zheng, MING Hong & CHEN Wei
ThermoFisher Scientific China, Shanghai 201206, China
With the realization of “the one hour yeast proteome”, in-deep proteome under rapid LC-MS/MS gradient is achieved. The current study utilized novel Q-OT-qIT mass spectrometer to analyze and optimize “the one hour proteome”. Within 50 min LC-MS/MS gradient, a total of 20860 and 14100 unique peptides were identified from 1 μg and 50 ng HeLa digest, corresponding to 3865 and 2877 proteins, respectively. Similar result from current reports used at least 2–3 h gradient. The effect of various collision modes, detectors, maximum injection times and automatic gain control settings were further evaluated, and the complementarity between different acquisition methods and scan parameters were described. Moreover, the massive parallelization of Q-OT-qIT was discussed, providing useful information for rapid proteome analysis.
proteomics, rapid identification, deep coverage, orbitrap, Q-OT-qIT MS
doi: 10.1360/10.1360/052014-16