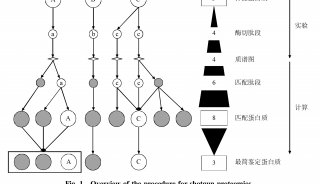
肽质量指纹图谱—MALDI-TOF 法鉴定蛋白质实验(二)
1. 目标样本沉积
1 ) 经典干燥液滴法
( 1 ) 半饱和氰基-4-羟基肉桂酸(约 5 mg/ml) 的配制:将 α-氰基-4-羟基苯甲酸溶解在 300 μl 1 : 1 乙腈/ TFA 溶 液(超声 10 min) 。离心几秒钟得到上清液。取 200 μl 上清液加到微量离心管中,并加入相同体积(200 μl ) 的 1 : 1 乙腈/TFA 溶液,就能配制成获得半饱和的 α-氰基-4-羟基肉桂酸。
( 2 ) 取 0.8 μl 消化溶液和 0.8 μl 基质加到微量离心管中,快速混合(避免在吸管尖结晶)。取 0.8 μl 混合液滴到 MALDI 标基上,其余的 0.8 μl 混合液可滴在 MALDI 标基的另一个位置。吸管尖头不要接触到标基。
( 3 ) 让混合液自然干燥并结晶出来(见注释 8) 。
2 ) 在疏水标基上干燥液滴法(见注释 9)
( 1 ) 准备 α-氰基-4-羟基肉桂酸:称取 10 mg 的 α-氰基-4-羟基肉桂酸,溶解在 1 ml 的 1:1 乙腈/TFA 溶液中(超声 10 min) 。取 56 μl 上述溶液放入一新的微量离心管中,并添加 994 μl 的 1 : 1 乙腈/ TFA 溶液。
( 2 ) 将 0.8 μl 凝胶消化后的上清液(见 19.3.1 节 3 ) 和 0.8 μl 上述溶液快速混合。
取 0.8 μl 混合液滴到 MALDI 标基上,其余的 0.8 μl 混合液可滴在 MALDI 标基的另一个位置 [ 见 19.3.2 节 1.1)]。
( 3 ) 让混合液自然干燥并结晶。
( 4 ) 滴加 4 μl TFA 溶液到晶体上,接触 30s 后,用吸管吸走液体(吸管尖不要接触晶体) 。
( 5 ) 加入 0.8 μl 的乙醇/丙酮/TFA 溶液,再次结晶。
( 6 ) 让混合液自然干燥并结晶出来。
2. 光谱采集
( 1 ) 将标基插入质谱仪中,并在真空中(低于 10-6 Torr) 稳定。
( 2 ) 打开髙压 [ 最好等待 20 min 让电压和温度恒定(焦耳效应 ) ]。
( 3 ) 调整激光功率(衰减)和标基位置,以便在 700~4000 Th 能得到好的信噪比和单一同位素分辨率。
( 4 ) 先激光发射 10 次,但所得图谱没有多大价值,因为它们通常是小质量分子( 基质和/或盐)的嘈杂图。
( 5 ) 激光发射 80~200 次,获取图谱并进行叠加。
( 6 ) 在 842.5099 Th 和 2211.1046 Th 处用水解胰蛋白酶得到离子进行质谱内部校正。
( 7 ) 保存图谱(见注释 10)。
3. 图谱解析
对单一同位素质量可以进行自动指认,但我们建议应仔细检查这些解析(见注释 11 和注释 12)。
如果没有对图谱进行平滑处理,可以对所有的初次单一同位素肽段峰进行解析,除了已知的胰蛋白酶产生的峰( 图 19-1,见注释 13 和注释 14 的细节描述)。
3.3 MASCOT 数据库检索使用并搜索结果的评价
下面描述的步骤是针对 MASCOT PMF 搜索引擎(Matrix Science,London, UK ) 内部授权(见注释 15 ) 或使用远在伦敦的服务器(http://www.matrixscience.com/cgi/search_ form, pi? SEARCH = PMF)。
当然,所述的搜索策略也可直接适用于其他搜索引擎(见注释 16)。
1. 搜索的第一步骤
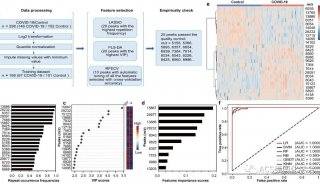
这个步骤(图 19-2 的左边)的目的是为了获得数据质量的总貌,以消除可能的污染物,并有可能直接确认植物蛋白质。
( 1 ) 确定一个全球开放的数据库(如 MSDB,NCBInr 或 SWISS- PROT) ,而不是特定物种之一,数据库包含有非植物的蛋白质污染物。
( 2 ) 不要在分类学领域(即 “ 所有项目” )指定品种,以确定非植物的蛋白质污染物。
( 3 ) 允许错过一个缺口。
( 4 ) 设置一个大的质量承受值为 100 ppm,以评估质谱校准(用于质谱校准的两个胰蛋白酶水解的肽段,它们的丰度和离子统计学意义可能很低,不能进行很好校准)。
( 5 ) 固定化修饰:羧甲基(C) 或脲甲基(C ) 。用碘乙酸或碘乙酰胺还原和烷基化凝胶样品的蛋白半胱氨酸残基,会分别产生羧甲基半胱氨酸或酰胺甲基半胱氨酸(包括在脲甲基半胱氨酸中)。
( 6 ) 变量的修改:无。
如果数据结果好(信噪比好,质量准确度高,污染物少)以及数据库中能搜索到参考的蛋白质,则可以得到希望的蛋白质信息(图 19-2B ) 。
候选蛋白质的评价标准有下面几条:
( 1 ) 分数优于重要临界值。
( 2 ) 最优蛋白质与非相关的蛋白质之间差别较大。
( 3 ) 所研究物种(或同源性很高的相近物种)的蛋白质为最优蛋白质第一级。
( 4 ) 候选蛋白质的分子质量和等电点与二维凝胶数据兼容。
( 5 ) 良好的质量准确度(最好)或不好的质量准确度(由于校准失败或错误校准问题 ) ,但在质量范围内,与质量分散具有低的和线性分布的关联。
( 6 ) —个裂解的肽段对三个匹配肽段的最大化。
( 7 ) 5 个匹配肽段的最小化。
( 8 ) 在蛋白质序列和序列覆盖中匹配肽段同质定位(见注释 17 和图 19-2C) 。
2. 第二步和后续搜索步骤
如果搜索没有得到结果,或者如果太多的峰(如超过 10 个)没有匹配(可能表明挖的点包含两个或更多的蛋白)(见注释 18 和图 19-2 的右边图),就需要进行进一步的搜索。
可选择更多的搜索条件(图 19-2D):
( 1 ) 删除确定非植物蛋白肽的消化污染物峰(“搜索无与伦比” 的吉祥物选项按钮 )。
( 2 ) 设置质量公差根据实际观察到的质谱的质量准确度(如作为评估非植物蛋白质污染物或核实有关胰蛋白质酶自动酶解片段)。通常情况下,优质的搜索要求质量准确度低于 30 ppm ( 较低值更好)。
( 3 ) 允许用非化学计量或不可预测的化学修饰搜索(主要是蛋氨酸氧化,天冬氨酸和谷氨酸甲基酯酰化,N 端焦谷氨酰甲基化(pyroglutamylation) ( 见注释 19) 。
( 4 ) 限制类群的搜索范围(如 Viridiplantae 蛋白质数据库)。
( 5 ) 允许在一个特定的数据库搜索。
在小节 3.3.1 中提及说明的评价和确认标准仍然很有必要(图 3-2E 和 F ) 。同时,鉴定混合物中次级组分也很重要。
通过肽质量指纹图谱(PMF ) 成功鉴定一个蛋白质后,在 MALDI-TOF 质谱中往往还有好几个离子峰不能鉴定出来。这些未鉴定出来的峰可能有不同的来源:
( 1 ) 在电泳凝胶中共同迁移的其他蛋白质的肽段离子峰。
( 2 ) 同一蛋白质的肽段离子峰,但是它的氨基酸序列和搜索数据库中的蛋白质序列有微小的差异(可能是基因组注释错误或突变引起的)。
( 3 ) 同一蛋白质的肽段离子峰,但是由于翻译后修饰的缘故,它的分子质量与预测的肽段质量不一致。
( 4 ) 因蛋白质污染出现的肽段离子峰(如角蛋白或胰蛋白)。
( 5 ) 因多聚体残渣或塑化剂污染出现的非肽段离子峰。
( 6 ) 因生物化学试剂药品污染出现的非肽段离子峰。
3.4 MALDI-TOF/TOF 策略
通过 PMF 鉴定的蛋白质只是作为初步的候选,鉴定蛋白质的得分情况表明可信程度。因此,我们应该进一步对 PMF 所得的离子峰进行二级测序,来提高并确认鉴定蛋白质的可信性。
在最近的 TOF/TOF 串联质谱分析仪未发明之前,在 MALDI 质谱分析仪上对肽段进行测序的成功率并不高。在 TOF/TOF 分析仪上,一级 TOF 能分离得到我们所需的肽段离子,其他肽段离子直接去掉,通过一级 TOF 的轰击筛选后,剩余的肽段都是特异的氨基酸序列肽段。然后二级 TOF 分离并测量这些特异氨基酸序列片段的分子质量。
1. 鉴定确认
为了确认鉴定的可信性,从单个蛋白质中鉴定的 1~3 个肽段通过 TOF/TOF 进行连续轰击。得到的片段离子必须和预测肽段序列相匹配。
2. 未知蛋白质的鉴定
在通过蛋白质数据库鉴定结果不理想的情况下( 见 3. 3) ,有些肽段(通常情况是 2~5 个)能通过 TOF/TOF 进行分析。从头开始测序结合蛋白质数据库搜索这些肽段离子的 TOF/TOF 离子片段,可能能成功鉴定这些未知蛋白质。搜索鉴定所选数据库应为研究对象所属分类或是种间交叉相关性的物种的蛋白质数据库(见第 20 章中LCMS/MS 部分)。