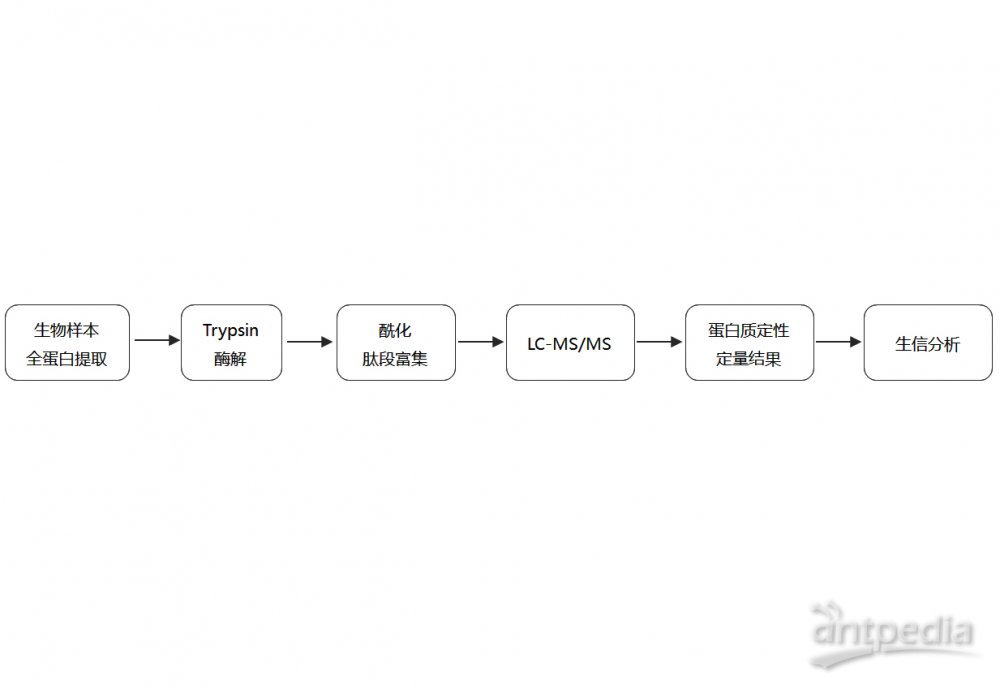
Dual- and Triple-Co... (二)
主要试剂
1. Blocking reagent after coating: 10% fetal calf serum (FCS) in phosphate-buffered saline (PBS), pH 7.4.
2. Cells suspended in complete culture medium, i.e., RPMI 1640 containing 10% FCS supplemented with glutamic acid, pyruvate, HEPES, and penicillin/streptomycin.
3. Monoclonal antibodies (mAbs) to cytokines: Mouse mAb to human IFN-γ; mAb 1-D1K for coating and FITC-labeled mAb 7-B6-1 for detection. Mouse mAb to human IL-2; mAbs IL2-I/249 for coating and biotinylated mAb IL2-II for detection. If a third cytokine is included in the analysis, e.g., IL-10, rat mAb 9D7 is used for coating and tag (different from FITC and biotin)-labeled rat mAb 12G8 is used for detection. All mAbs from Mabtech, Nacka Strand, Sweden.
4. Secondary reagents for amplification: Mouse anti-FITC mAb labeled with green fluorophore emitting light at the same wavelength as FITC, streptavidin (SA) labeled with Cy3, and if three cytokines are analyzed, a mouse anti-tag mAb labeled with Cy5 (Mabtech).
5. Mouse mAb anti-human CD28 at 0.1 mg/ml in sterile filtered (0.2 μm) PBS (Mabtech).
主要设备
1. Transparent, flat-bottomed, 96-well plates with polyvinylidene difluoride (PVDF) membranes designed for low autofluorescence (IPFL; Millipore, Cork, Ireland).
2. Analysis of spots is performed with an iSpot Spectrum FluoroSpot Reader from AID Diagnostika GmbH, Strassberg, Germany, equipped with a motorized stage, a digital firewire camera of 5.2 MP, and a 7&1 filter wheel equipped with narrow band filters for DAPI, FITC, Cy3, and Cy5.
3. Fluorescence Enhancer (Mabtech) is used to improve the fluorescent signal.
实验步骤
The protocol describes double staining for the detection of human IFN-γ and IL-2. PVDF plates are coated with mAbs to IFN-γ and IL-2. Cells ± stimuli are added, and secreted IFN-γ and IL-2 are captured by the specific mAbs. After cell removal, spots are detected in two steps. First, a mixture of mAb anti-IFN-γ-FITC and anti-IL-2-biotin is added. Secondly, spots are detected by adding a mixture of mAb anti-FITC-green fluorophore (for IFN-γ) and SA-Cy3 (red fluorescence; for IL-2).
The protocol can be adapted to triple staining by using three coating antibodies, three detection antibodies labeled with different tags, and finally three different antitag amplification reagents labeled with different fluorophores (green, Cy3 and Cy5). The protocol for double staining is well-established, whereas the protocol for and analysis of triple staining have to be further optimized. The protocol can also be adjusted to single staining by using reagents for only one cytokine.
1. Dual-Color Fluorospot
1) Preparation of the plates and coating for double staining (sterile conditions)
a. Pre-wet a sterile 96-well PVDF plate with 20 μl of 35% ethanol for 1 min at room temperature and wash the plate five times with 200 μl of sterile water per well.
b. Mix coating mAbs for IFN-γ and IL-2 in the same tube in sterile PBS, pH 7.4, at a final concentration of 15 μg/ml each. Add 100 μl/well of the mixed mAb solution and incubate overnight at 4–8°C (seeNote 1).
c. After coating, empty the plate and wash five times with sterile PBS (200 μl/well) to remove excess mAb. Block the plate for at least 30 min with 200 μl/well of 10% FCS (same serum as used for the cell suspensions) in PBS (seeNote 2).
2) Incubation of cells in the plates (sterile conditions)
a. Suspend the cells in complete culture medium (seeNote 3) and add the suspension, including stimulatory agents of choice and anti-CD28 (0.1 μg/ml; seeNote 4), in a final volume of 100–150 μl/well. The number of cells per well is dependent on the stimulating agent used (seeNote 5).
b. Incubate the plate overnight at 37°C in a humidified atmosphere with 5% carbon dioxide. It is important to wrap the plate in aluminum foil to avoid evaporation and to avoid moving the plate during this time. Incubation times may vary depending on the cytokine to be detected (seeNote 6).
3) Detection of spots
a. Discard the cells and wash the plate five times with PBS in a plate washer (or by hand 200 μl/well; seeNote 7).
b. In the same tube, dilute the detection mAbs for IFN-γ and IL-2 labeled with FITC and biotin, respectively, in PBS containing 0.1% bovine serum albumin (BSA) (seeNote 8). Add 100 μl/well and leave the plate for 2 h at room temperature.
c. Wash the plate as in step 6 above.
d. Dilute secondary detection reagents, i.e., mAb anti-FITC labeled with green fluorophore and SA labeled with Cy3, in the same tube to their respective working concentrations in PBS–0.1% BSA (seeNote 8). Add 100 μl/well and leave the plate for 1 h at room temperature.
e. Wash the plate as in step 6 above, tap the plate against tissue paper, and add thereafter fluorescence enhancer solution for 15 min at room temperature (100 μl/well). Finally, tap the plate again against clean tissue paper. Remove the soft plastic underdrain and let the plate dry in the dark before analysis. Further storage of the dry plate should be at room temperature in the dark.
2. Analysis of Spots
1) The plate should be completely dry before analysis. We recommend the use of an automated reader with filters for FITC and Cy3. Green spots represent IFN-γ-producing cells and red spots IL-2-producing cells. Double-stained spots are then identified based on the position of the green and red spots in a computerized overlay of IL-2 and IFN-γ spot images from the same well. Similarly, in triple staining, the reader has to be able to identify and analyze yet another fluorophore, in this case Cy5.
2) Fluorescent spots, like any fluorescent signal, may fade due to excessive exposure to light. Although the fluorescence in the dry plate has been found to be surprisingly stable (plates can be saved for months), for best result it is recommended to analyze the plate within 1 week of development as some brightening of the membrane, especially in green staining, takes place with time.
3) To ensure that no “bleeding over” between filters occurs, wells where only one detection system has been used can be included; analysis with filters for the other fluorophore should result in an image with no detectable spots.
4) Random dual spots may occur, i.e., two adjacent cells secreting either of the two cytokines of interest may be counted as one dual spot. The number of random dual spots is primarily dependent on the total number of spots of each cytokine in the well and the definition criteria for multistained spots set in the reader (maximal distance between spot centers). If this distance is set to about 15 μm, experiments in our laboratory have shown that 100 spots of each cytokine per well will generate less than 1% random events. Similarly, at about 500 spots of each cytokine per well, the percentage of random events does not exceed 2%. One way to tentatively estimate the number of random spots in any given experiment is to analyze the number of dual spots in overlays of unrelated green and red images from different wells within, e.g., a triplicate (12). In a similar manner, different combinations of random double-positive spots can be estimated in triple staining experiments. Also random triple-positive spots, which occur at a very low frequency, can be estimated in this way.
注意事项
1. Plates: Transparent plates with low-fluorescent PVDF-based membrane are recommended. To obtain maximal antibody binding capacity, these plates need to be pre-wetted with ethanol as described. It is essential to keep the membrane wet after ethanol treatment. If the membrane is let to dry, the pre-wetting needs to be repeated before addition of the coating antibodies. To avoid artifactual staining, working antibody solutions should preferably be filtered (0.2 μm).
2. Blocking: Lower membrane autofluorescence is obtained if blocking is performed with PBS containing 10% FCS instead of complete medium.
3. Serum in cell medium: A serum pre-evaluated for cell culture and that yields low spontaneous cytokine secretion should be used in the medium; FCS is recommended.
4. Effect of IL-2 capture and costimulation with anti-CD28: When IFN-γ and IL-2 are measured in the same well, capture of secreted IL-2 by coated mAbs to IL-2 may have a negative impact on the production of IFN-γ and other cytokines (13). A proper control for that potential effect is to include wells coated with a single capture mAb. Costimulation using anti-CD28 mAb enhances IL-2 production which can restore IFN-γ production to the same level as when IFN-γ is analyzed separately. Inclusion of anti-CD28 in the cell suspension added to double-coated wells is recommended. Further optimization may be necessary, depending on which cells and stimuli are used. A too high concentration of anti-CD28 mAb may result in elevation of nonspecific cytokine secretion, in particular IL-2. The costimulatory effects of anti-CD28 mAb, as well as a possible impact on nonspecific spots, can be assessed by comparing cells cultured with or without anti-CD28 mAb. An alternative strategy to avoid IL-2 capture effects is to preincubate the cells in vials prior to adding them to the coated wells. In this case, the cells are activated before they are exposed to IL-2 capture antibodies.
5. Cells: Both freshly prepared and cryopreserved cells may be used in the assay with good results. Triplicates or duplicates of 250,000 peripheral blood mononuclear cells (PBMCs) per well are often used to assess antigen-specific T-cell responses. If cryopreserved cells are used, they should be rested for a minimum of 1 h at 37°C in 5% CO2 before being counted and mixed with stimulatory agents. The number of cells responding to antigen stimulation is often compared to the number of cells spontaneously producing cytokine. Spontaneous production is determined by incubating the same number of cells in the absence of antigen. A polyclonal activator, such as anti-CD3 mAb (100 ng/ml) or phytohemagglutinin (PHA; 1–10 μg/ml), should normally be used as a control for cell viability and functionality of the test system. For polyclonal activators, the number of PBMC per well may have to be reduced in order to avoid confluent spot formation. Typical cell numbers for stimulation with anti-CD3 or PHA are 50,000–100,000 PBMC per well. If T-cell clones, mixtures of separated cell fractions, etc. are used, conditions may have to be modified. In situations where most cells secrete the cytokine of interest, i.e., cell lines, the number of cells per well should be even further reduced.
6. Kinetics: When two cytokines are investigated, the cell incubation time has to be adapted to fit both. In T-cell responses manifested by IFN-γ and IL-2 production, both cytokines are rapidly induced and can be detected already after 10 h. In cases where the cytokines have different kinetics, e.g., analysis of IFN-γ together with a cytokine like IL-4, the incubation time may have to be extended. For triple staining, the kinetics of all three cytokines have to be considered.
7. Plate washing: Washing of plates can be done using a multichannel device. In washing steps not requiring sterile conditions (from “Detection of spots”, step 6 and forward), a regular ELISA plate washer with a washing head adjusted to ELISPOT/fluorospot plates can be used.
8. Concentration of detection reagents: Suitable working concentrations for both the primary and secondary detection reagents should be determined in pilot tests. In the double-staining protocol described, working concentrations for the respective reagents were mAb anti-IFN-γ-FITC (1 μg/ml), mAb anti-IL-2-biotin (2 μg/ml), mAb anti-FITC-green (1 μg/ml), and SA-Cy3 (0.5 μg/ml).