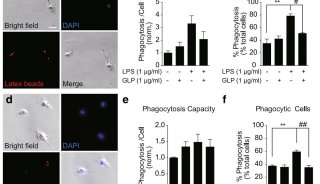
常用细胞凋亡检测方法(图)-3
过氧化物酶标记测定法
原理:脱氧核糖核苷酸衍生物地高辛[(digoxigenin)-11-dUTP]在TdT酶的作用下,可以掺入到凋亡细胞双链或单链DNA的3-OH末端,与dATP形成异多聚体,并可与连接了报告酶(过氧化物酶或碱性磷酸酶)的抗地高辛抗体结合。在适合底物存在下,过氧化物酶可产生很强的颜色反应,特异准确的定位出正在凋亡的细胞,因而可在普通光学显微镜下进行观察毛地黄植物是地高辛的唯一来源。在所有动物组织中几乎不存在能与抗地高辛杭体结合的配体,因而非特异性反应很低。抗地高辛的特异性抗体与脊椎动物甾体激素的交叉反应不到1%,若此抗体的Fc部分通过蛋白酶水解的方法除去后,则可完全排除细胞Fc受体非特异性的吸附作用。本方法可以用于福尔马林固定的石蜡包埋的组织切片、冰冻切片和培养的或从组织中分离的细胞凋亡测定。
一、试剂配制
1、磷酸缓冲液PBS(pH7.4):磷酸钠盐 50mM,NaCl 200mM。
2、蛋白酶K(200μg/ml,pH7.4):蛋白酶K 0.02g;PBS 100ml。
3、含2%H2O2的PBS缓冲液(pH7.4):H2O2 2.0ml;PBS缓冲液 98.0ml。
4、TdT酶缓冲液(新鲜配):Trlzma碱 3.63g用 0.1N HCl调节pH至 7.2,加ddH2O定容到1000ml;再加入二甲砷酸钠 29.96g和氯化钴0.238g。
5、TdT酶反应液:TdT酶32μl;TdT酶缓冲液 76μl,混匀,置于冰上备用。
6、洗涤与终止反应缓冲液:氯化钠 17. 4g;柠檬酸钠 8.82g;ddH2O 1000ml
7、0.05%二氨基联苯(DAB)溶液:DAB 5mg;PBS 10ml,pH7.4, 临用前过滤后,加过氧化氢至0.02%。
8、0.5%甲基绿(pH4.0):甲基绿0.5g;0.1M乙酸钠100ml。
9、100%丁醇,100%、95%、90%、80%和70%乙醇,二甲苯,10%中性甲醛溶液,乙酸,松香水等。
10、 过氧化物酶标记的抗地高辛抗体(ONCOR)
二、实验步骤
1、标本预处理:
(1)石蜡包埋的组织切片预处理:将组织切片置于染色缸中,用二甲苯洗两次,每次5min。用无水乙醇洗两次,每次3min。用95%和75%乙醇各洗一次,每次3min。用PBS洗5min 加入蛋白酶K溶液(20ug/ml),于室温水解15min,去除组织蛋白。用蒸馏水洗4次,每次2min,然后按下述步骤2进行操作。
(2)冰冻组织切片预处理:将冰冻组织切片置10%中性甲醛中,于室温固定10min后,去除多余液体。用PBS洗两次,每次5min。置乙醇:乙酸(2:1)的溶液中,于-20℃处理5min,去除多余液体。用PBS洗两次,每次5min,然后按下述步骤2进行操作。
(3)培养的或从组织分离的细胞的预处理:将约 5 × 107个/ml细胞于 4%中性甲醛室温中固定 10min。在载玻片上滴加 50~100μl细胞悬液并使之干燥。用PBS洗两次,每次5min,然后按下述步骤2进行操作。
2、色缸中加入含2%过氧化氢的PBS,于室温反应5min。用PBS洗两次,每次5min。
3、用滤纸小心吸去载玻片上组织周围的多余液体,立即在切片上加2滴 TdT酶缓冲液,置室温1~5min。
4、 用滤纸小心吸去切片周围的多余液体,立即在切片上滴加 54μl TdT酶反应液,置湿盒中于37C反应 1hr(注意:阴性染色对照,加不含TdT酶的反应液)。
5、将切片置于染色缸中,加入已预热到37℃的洗涤与终止反应缓冲液,于37℃保温30min,每10min将载玻片轻轻提起和放下一次,使液体轻微搅动。
6、 组织切片用PBS洗 3次,每次 5min后,直接在切片上滴加两滴过氧化物酶标记的抗地高辛抗体,于湿盒中室温反应30min。
7、用PBS洗4次,每次5min。
8、在组织切片上直接滴加新鲜配制的0.05%DAB溶液,室温显色3~6min。
9、用蒸馏水洗4次,前3次每次1min,最后1次5min。
10、于室温用甲基绿进行复染10min。用蒸馏水洗3次,前两次将载玻片提起放下10次,最后1次静置30s。依同样方法再用100%正丁醇洗三次。
11、用二甲苯脱水3次,每次2min,封片、干燥后,在光学显微镜下观察并记录实验结果。
三、注意事项
一定要设立阳性和阴性细胞对照。阳性对照的切片可使用DNaseI部分降解的标本,阳性细胞对照可使用地塞米松(1μM)处理3-4hr的大、小鼠胸腺细胞或人外周血淋巴细胞。阴性对照不加TdT酶,其余步骤与实验组相同。
丁香网友医琳师妹所作的PC12细胞培养方法以及诱导凋亡的方法:
PC12细胞培养方法
一、器具:
1、培养皿(35mm)(或50cm2培养瓶)
2、6孔(35mm)培养板、24孔培养板
3、滴管
4、小青瓶
5、100ml、250ml玻璃瓶
6、翻口橡皮塞
7、烧杯:1000ml、500ml、100ml、50ml
8、容量瓶:1000ml、500ml、100ml
9、量筒:50ml、100ml、250ml
10、玻璃棒
11、pH试纸
12、滤器(0.22μl,γ射线菌一次性使用)
13、注射器:10ml、20ml
14、24mm×24mm盖玻片
二、器具处理:
玻璃器皿、塑料器皿,予清洗、泡酸、三蒸水冲洗,干燥后备用。然后高压蒸气灭菌,置超净台紫外线灯照,风机抽干。
三、试剂:
1、培养基:DMEM(高糖)(或RPMI-1640)
2、马血清(56℃,30分钟灭活)
3、胎牛血清(56℃,30分钟灭活)
4、青霉素
5、链霉素
6、NaHCO3
7、HCl
8、谷胺酰胺(选用)
9、神经生长因子(选用)
10、磷酸二氢钠
11、磷酸氢二钠
12、氯化钠
13、多聚甲醛
14、胰蛋白酶
15、NaOH(选用)
16、多聚赖氨酸(或鼠尾胶)
17、D-Hank’s液
18、乙醇(无水、75%、95%)
四、配试剂:
1、DMEM不完全培养基:
DMEM
Hepes 3.08g
NaHCO3 3.7g
100U/ml青霉素/100mg/L链霉素
(调PH至7.2-7.4),不加血清为不完全培养基。
2、100U/ml青霉素/100mg/L链霉素:
青霉素80万U+NS→40ml
链霉素100mg/L+NS→50ml取40ml
混成80ml,抽滤分装,4℃保存。
3、DMEM完全培养液
DMEM
10%胎牛血清(已灭活)
5%马血清
调pH7.2-7.4
加100U/ml青霉素、100mg/L链霉素约1ml
4、RPMI1640不完全培养液:
RPMI1640,25mM HEPES,用5%NaHCO3调节pH至7.4,灭菌后4℃保存,即:
RPMI-1640 10.4g
Hepes 3.58g
三蒸水 1000ml
1N NaHCO3(或NaOH)调pH值至7.2-7.4抽滤分装90ml/瓶,4℃或-20℃冻存。
5、RPMI1640完全培养液:
25mM HEPES,5μg/ml胰岛素,5μg/ml转铁蛋白,100U/ml青霉素,100 mg/ml链霉素及20%FCS,用5%NaHCO3调节pH至7.4,灭菌后4℃保存。
RPMI-1640不完全培养液,加100U/ml青霉素/100mg/ml链霉素,临用前加入10%或20%FCS(v/v,胎牛血清),用1N NaHCO3调节pH值7.4,抽滤灭菌后4℃保存。
6、0.25%胰蛋白酶(DMEM配)
胰蛋白酶250mg
DMEM 100ml
抽滤分装小青瓶,-20℃冻存
7、0.25%胰蛋白酶(RPMI 1640配)
胰酶250mg,加RPMI 1640 100ml,用0.1N NaHCO3调节pH至7.4,灭菌后4℃保存。
8、 Hank’s平衡盐溶液:5.4mM KCl, 0.3mM Na2HPO4, 0.4mM KH2PO4, 4.2mM NaHCO3, 1.3mM CaCl2, 0.5mM mgCl2, 0.6mM MgSO4, 137mM NaCl, 5.6 mM D-葡萄糖,0.02%(w/v)酚红。
9、0.1M pH7.4 PBS:
137mM NaCl, 2.7mM KCl, 4.3mM Na2HPO4•7H2O, 1.4mM KH2PO4。
10、1%多聚甲醛:多聚甲醛1g,加0.01M pH7.4 PBS至100ml。
11、抗体稀释缓冲液:0.01M PBS, 0.1%BSA, 0.1Na3N。
12、小牛血清:
56℃,灭活30分钟
分装小青瓶,10ml或20ml/瓶
13、马血清:
56℃,灭活30分钟
分装小青瓶,10ml或20ml/瓶
14、0.1M PBS:
137mM NaCl
2.7mM KCl
4.3mM Na2HPO4•7H2O
1.4mM KH2PO4
15、7.4%NaHCO3
NaHCO3 7.4g
双蒸水 100ml
抽滤分装小青瓶,4℃保存。
16、0.1N HCl
1N HCl 10ml
双蒸水 100ml
抽滤分装小青瓶,4℃保存。
17、多聚赖氨酸(包被用):
多聚赖氨酸氢溴化物溶解于150m mol/L硼酸钠缓冲液中,(pH8.4),终浓度为1mg/ml,抽滤灭菌。(玻璃器皿可用50μg/ml多聚-L赖氨酸氢溴化物的水溶液。)
18、神经生长因子:
用pH5.0的醋酸钠缓冲液(0.1mol/L)配置NGF(2.5S)250μg/ml贮存液,使用前再用培养液稀释,贮存液的浓度太低,活性容易丧失,贮存液应冻存于-80℃,一旦冻融后不宜再冻存,所以应将其分装成小份冻存,冻融后的NGF贮存液可分装于EP管中,置于4℃贮存,其活性可保存数月。
五、操作步骤:
1、将多聚-L-赖氨酸氢溴化物(终浓度1mg/ml),经过滤除菌后用于包被培养皿,在室温作用4小时后,吸去包被溶液,并将培养皿用无菌培养用水漂洗4次,置超净台里晾干。(可再予紫外线照射20分钟灭菌)。(对玻璃器皿,可将经过滤除菌的多聚-L-赖氨酸氢溴化物水溶液50μg/ml加于玻璃器皿表面包被1小时,然后用无菌培养用水漂洗4次,并置超净台内空气干燥。
2、将PC12细胞(2×104或2×105)接种于含10%胎牛血清、5%马血清, 100U/ml青霉素、100mg/ml链霉素的DMEM完全培养液(pH7.2)的塑料培养瓶(或培养皿)中。
3、置37℃、5%CO2细胞培养箱中培养。
4、细胞接种后,每2-3天换液一次。每次更换约2/3的培养液,并在倒置显微镜下观察PC12细胞是否贴壁。
5、80-90%融合时,用0.25%胰蛋白酶溶液消化,D-Hank’s液收集细胞,调整细胞数。转种到6孔培养板中(可以先用多聚赖氨酸包被)。接种密度为5×105/ml.孵育48h后作为实验细胞。(有人每周按1:4传代)。如准备用于激光共聚焦显微镜照相,可先将处理过的24mm×24mm盖玻片,再转种细胞到6孔培养板中。
6、吸去培养液,用D-Hanks液洗2遍,每孔加入含干预药物的无血清DMEM不完全培养液适当时间,可以进行测定增殖、凋亡或细胞周期情况。
7、简述:(南医大神经药理)将PC12细胞接种于培养液中,培养液为含10%胎牛血清的DMEM培养液,37℃,5%CO2条件下培养,待细胞长满瓶底后,倾去培养液,用D-Hank’s液轻轻荡洗两次,用吸管轻轻吹打数次,使细胞完全分散,倒置显微镜下观察计数,调整细胞浓度为:5×105个/ml,接种于24孔培养板,待细胞长成连续单层后,予干预。
8、神经生长因子诱导PC12细胞分化神经纤维:要求:(1)细胞密度: 2.5-10×104 cell/cm2(2)塑料培养皿的包被。上述细胞培养24小时后,换入含2.5S NGF(50ng/ml)的DMEM完全培养液,以后隔天换液(含NGF);取NGF分化5-10天后的PC12细胞用于实验。(一般情况下,NGF诱导10-14天时,NGF可使PC12细胞长出神经纤维,并形成致密的网络)。
9、PC12细胞凋亡模型的制备与干预:(1)无血清诱导:PC12细胞培养后用无血清的DMEM洗三次,加无血清的DMEM培养12小时,然后予各种干预。最后予4℃预冷的75%乙醇固定后,送流式细胞仪以PI(碘化丙啶)检测。(2)谷氨酸诱导:配置含80μM-10mM终浓度的谷氨酸(经典用10mM)的DMEM不完全培养液(不含血清),处理24小时(6-24小时,可偏长一点)后,再予各种干预,最后送流式细胞仪测定细胞凋亡(有人要求在无血清培养基中先培养8-24小时后,再加含10mM谷氨酸的不完全DMEM培养液培养6-24小时。)(3)缺氧诱导:首先设置低氧环境,将培养箱内充入95%氮气代替空气,同时充入5%二氧化碳,条件稳定后(5%二氧化碳:5%氧气),将细胞置入培养,分别培养30分钟、1小时、2小时、3小时、6小时、12小时,取出细胞,将细胞吹打成细胞悬液,1000r/min离心30分钟,倾去上清,PBS洗涤一次,离心去上清后,迅速注入3ml 4℃70%的冷乙醇,边注入边振摇,同时反复通过7号针头的注射器抽打5次(或用振匀器振匀),以避免细胞成团,送流式细胞仪PI检测。
10、吖啶橙/溴化乙锭双荧光染色测定细胞凋亡(acridine orange/ethidium bromide, AO/EB):(细胞爬片),在6孔培养板中预先置入玻璃盖玻片,接种细胞悬液,干预后,予95%乙醇固定15分钟,微干,然后准备荧光染色。将100mg/L溶于PBS的吖啶橙和100mg/L溶于PBS的溴化乙锭各5μl(临用前混合加入,加样量宜小,有时各2μl-5μl足够,量太大容易形成细胞凋亡的假阳性),在照相前混匀后加入,轻轻吹打,30秒后用激光共聚焦显微镜观察照相。(有人用15mg吖啶橙溶于100ml pH6.8的PBS中过滤,避光保存。)计取5个视野,共100个细胞中凋亡细胞百分数。(如制成细胞悬液,取100μl PBS稀释的细胞悬液,加2μl的染色液,其中AO/EB,各含100μg/ml,加入染料30秒后观察摄像。)(有人用PBS配制吖啶橙/溴化乙锭(AO/EB)混合液,各含100μg/ml。细胞离心,悬液于PBS中,取100μl细胞悬液加4μl荧光染色液,轻轻吹打,滴至载玻片上,加盖玻片后立即置荧光显微镜下观察。)
-
科技前沿

-
焦点事件

-
焦点事件

-
焦点事件