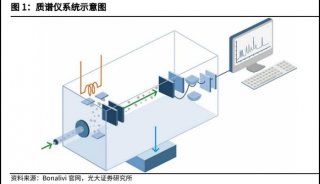
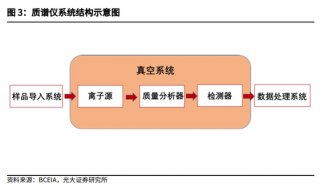
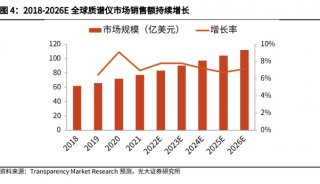
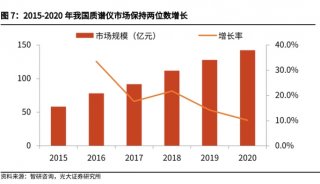
苯丙酮尿症(PKU)的筛查与治疗
苯丙酮尿症(Phenylketonuria,PKU)是最常见的氨基酸代谢遗传性疾病之一,苯丙酮尿症(PKU)是为数不多的可治性、遗传性、代谢性疾病,能通过限制苯丙氨酸的摄入量进行治疗。本文对苯丙酮尿症(PKU)的筛查与治疗情况进行回顾和综述,以期提高此病的诊断率,使患有苯丙酮尿症及高苯丙氨酸血症的患者能从早期治疗中获益。
1 苯丙酮尿症(Phenylketonuria,PKU)的发病机理
苯丙酮尿症(PKU)属常染色体隐性遗传病,系因苯丙氨酸羟化酶(Phenylketonuria
hydrox-2-lase,PAH)基因突变,导致PAH活性降低或丧失,苯丙氨酸(Phenylalanine,Phe)在肝脏中代谢紊乱所致。正常情况下苯丙氨酸代谢的主要途径是通过肝细胞中PAH转化为酪氨酸,以合成甲状腺素、肾上腺素和黑色素等。苯丙酮尿症(PKU)患者由于缺乏PAH,Phe不能羟化为酪氨酸,而循另一条代谢通路,即Phe与α-酮戊二酸进行转氨基作用生成苯丙酮酸,大量的苯丙酮酸在血液与组织中堆积并排泄于尿液中,形成苯丙酮尿症(PKU)。截止2003年,已有超过400种不同位点的PAH基因突变被证实,其中仅在欧洲就有29种典型的PAH基因突变类型,且分布呈地区性。我国学者对北京地区苯丙酮尿症(PKU)患儿的PAH基因突变的研究提示常见的突变有R243Q/EX6-96A>G/Y356X等。
目前认为我国北方PAH基因突变的类型与亚洲其他地区相似,但与欧洲人群有明显的差异。Phe转化为酪氨酸的过程中除需PAH外,还必须有四氢生物蝶呤(tetrahydrobiopterin,BH4)作为辅酶参与。人体内的BH4是由鸟苷三磷酸(guanosine
triphosphate,GTP),经过鸟苷三磷酸环化水合酶(guanosine triphosphate
cyclohydrase,GTP-CH)、6-丙酮酸四氢蝶呤合成酶(6-pyruvic acid tetrahydrobiopterin
synthetase,PTPS)和二氢生物蝶呤还原酶(dihydrobiopterin
reductases,DHPR)等一系列酶的催化而合成,PAH、GTP-CH、DHPR三种酶的编码基因分别定位于12q24.1、14q11,4p15.1-p16.1;而对PTPS编码基因的研究尚在进行中,上述任一编码基因的突变都有可能造成相关酶的活性缺陷,致使苯丙氨酸发生异常累积,亦可发生苯丙酮尿症(PKU)。
2 苯丙酮尿症(PKU)的临床表现
2.1 智力低下
智力低下是本病的最主要表现。患儿出生时正常,生后数月出现呕吐、易激惹、生长迟缓等现象,未经治疗者在4~9个月就出现明显的智能发育落后,语言发育障碍尤甚。半岁以内智商多在90左右,半岁后迅速降低,1岁后在50左右,3岁后在40左右。智力低下的程度不同,约60%属于重型低下(IQ<50),余为中、轻型,仅有1%~4%的病例智力接近于正常(IQ≥80)。
2.2 惊厥
约1/4患儿有癫痫发作,常在生后18个月发作,多见于有严重智力低下者。发作类型可为婴儿痉挛、全身强直阵挛发作或部分性发作。约80%的患儿有脑电图异常。癫痫发作可随年龄的增长逐渐减轻或自动停止,有的变换发作形式。
2.3 神经、精神症状
常见精神行为异常,如兴奋不安、多动攻击性行为或孤独样行为。神经系统体征不多见,约1/3患儿有不典型体征,如肌张力增加、步态异常、腱反射亢进、手部轻微震颤、肢体的重复动作等,严重的可出现脑性瘫痪。
2.4 色素脱失症
约90%的患儿表现毛发变红、皮肤发白、虹膜色浅,但不像白化症出生时就存在,一般是出生后至半岁左右逐渐显著,1岁左右毛发呈黄色或金发,色素缺乏的程度也不像白化症那样完全。
2.5 其他症状
约1/3患儿皮肤干燥,易生湿疹,且对一般治发无效。患儿有特殊的发霉样气味或鼠尿味。值得指出的是,苯丙酮尿症(PKU)患者往往同时出现上述几个症状,与其他神经系统疾病容易混淆。夏静[4]等报道1例苯丙酮尿症(PKU)患儿1岁后出现皮肤、毛发异常,智力发育迟缓,误诊为脑性瘫痪。Guillemette
Jousserand等报道一例54岁的苯丙酮尿症(PKU)患者表现出鼠尿样气味、四肢麻痹,轻度瘫痪并伴有认知缺损等症状。可见上述症状也不仅局限于婴幼儿。
3 苯丙酮尿症(PKU)的临床筛查与检验
因苯丙酮尿症(PKU)患儿在2个月内没有明显症状,当临床症状明显时婴儿的智力发育等已受到不可逆的损伤,且与其他神经系统疾病的症状容易混淆,所以加强苯丙酮尿症(PKU)的临床筛查和检验就显得尤为重要。《新生儿疾病筛查管理办法》(卫计委令第64号)将苯丙酮尿症(PKU)的筛查立为新生儿必须筛查的遗传代谢病之一。目前国内针对苯丙酮尿症(PKU)的筛查检验方法主要有:
(1)化学荧光分析法。婴儿出生72h后吃饱高蛋白奶6次以上,针刺足跟内采血,滴于采血滤纸上自然干燥,采用化学荧光分析法测定血清内Phe的浓度,≥1200μmol/L则诊断为苯丙酮尿症(PKU)。
(2)Phe负荷试验。患儿口服苯丙氨酸100ng/kg,服用前及服用后1、2、3、4h分别测定血清Phe浓度;或人工喂养高蛋白奶粉3天后测定血清Phe浓度,大于或等于1200μmol/L则诊断为苯丙酮尿症(PKU)。
(3)PCR检测。金煜炜等建立了联合应用PCR-单链构象多态性和短串联重复序列连锁分析方法产前诊断苯丙酮尿症(PKU)患儿,初步结果表明该方法准确性高,具有较高的临床应用价值。目前对苯丙酮尿症(PKU)患者的筛查和临床检验已经达到基因水平,如单链多态性PCR检测、高压液相色谱定量(HPLC)法等已经应用于临床,为苯丙酮尿症(PKU)的早期诊断和早期治疗提供检验学依据和基础。
4 苯丙酮尿症(PKU)的治疗
Giovannini M等人提出,治疗苯丙酮尿症(PKU)的主要原则是:
(1)给患儿提供适量的苯丙氨酸维持其正常成长;
(2)保证摄入足够其他营养素,如长链多不饱和脂肪酸,使患儿能够保持良好的营养状态;
(3)保证患儿对治疗的最佳依从性;
(4)同时考虑到患儿的生活质量。
目前国内外对苯丙酮尿症(PKU)的治疗主要有以下几种。
4.1 限制苯丙氨酸摄入的传统饮食疗法
由于患儿体内无法正常代谢苯丙氨酸,所以必须限制摄入,防止异常代谢产物在体内积聚至毒性水平。另一方面,苯丙氨酸属于必需氨基酸,缺乏了会影响患儿的发育,患儿的主要饮食是低蛋白的不含苯丙氨酸的氨基酸混合物,同时加入矿物质、维生素和其他营养素,可见苯丙酮尿症(PKU)患者的饮食控制是非常严格的。饮食疗法对于婴幼儿患者是必要的,但是随着患者年龄的增长,其依从性往往下降,有报道长期坚持苯丙氨酸控制饮食的患儿也会表现出大脑功能受损,智力发育低于同等年龄儿童,所以尽管控制苯丙氨酸饮食对苯丙酮尿症(PKU)有效,但并不是一种理想的治疗方法。
4.2 BH4相关药物治疗
如前所述,Phe的代谢除了受苯丙氨酸羟化酶影响外,也受到BH4的影响,前者缺乏即为典型苯丙酮尿症(PKU),而后者的异常则表现为四氢生物蝶呤异常型。有学者发现,某些PAH缺乏的苯丙酮尿症(PKU)患者在补充BH4后血浆内Phe的水平有所降低,而BH4反应性的苯丙酮尿症(PKU)患者单独服用BH4也可取得良好的效果。目前的商品化的BH4为二盐酸沙丙蝶呤片(sapropterin
dihydrochloride),于2007年12月13日获得美国FDA批准上市,商品名为Kuvan。大约20%的苯丙酮尿症(PKU)患者对沙丙蝶呤的治疗有反应,导致血苯丙氨酸浓度的下降。而在另一个研究中,采用双盲的方法对两组患者进行检测,6周后,95%的实验组患者和9%的对照组患者的血苯丙氨酸浓度下降了至少30%。提示某些对BH4反应的患者可以应用沙丙蝶呤作为治疗方法补充饮食治疗,甚至可能替换饮食治疗。但Alberto
Ponzone等的实验表明大多数苯丙酮尿症(PKU)患者服用BH4制剂后血浆内Phe和酪氨酸的水平与对照组无明显差异,他们认为BH4在苯丙酮尿症(PKU)患者蛋白代谢途径中的作用还有待于进一步研究。
另外,有些药物还在实验阶段。如苯丙氨酸解氨酶(phenylalanine ammonia-lase,
PAL),该酶存在于肝脏中,负责饮食中部分苯丙氨酸的降解,其降解产物氨和转苯乙烯酸对苯丙酮尿症(PKU)患者无害。PAL经过固定后可永久地降解苯丙氨酸进而抑制其吸收,动物实验提示其降低苯丙氨酸水平的能力甚至高于饮食控制。其缺点是口服易被肠道内的消化酶分解,而注射又有较强的免疫原性。目前,科学家正在采用体外重组的方法合成PAL并用于动物实验。目前针对PAH缺乏的基因治疗方法也在研究中。科学家们将携带表达PAH基因的cDNA的重组腺病毒放置入PAH缺陷小鼠的肝循环,10%~80%的肝脏PAH活性得以恢复,进而保持血清中的苯丙氨酸浓度正常。这一方法的主要问题是转运效率低和抗重组腺病毒载体的抗体出现。科学家们试图应用免疫抑制剂减少干扰腺病毒载体的抗体出现,并在小鼠实验中取得了一定疗效。我们期待今后的几年里,应用一种或多种治疗苯丙酮尿症(PKU)的方法,结合不很严格的饮食限制,完全控制苯丙酮尿症(PKU)患者血苯丙氨酸的水平。然而,在婴儿期,低苯丙氨酸饮食仍是最优选择。但一旦患者难以继续坚持饮食治疗,应该采取一些替换治疗。
5 小结
苯丙酮尿症(PKU)属于基因水平的疾病,也是少数可以治愈的疾病,如果能够开展大规模的病例筛查,做到早诊断、早治疗,就能最大限度地减轻苯丙酮尿症(PKU)患者的临床症状,保证其正常生长发育,提高生活质量。目前针对苯丙酮尿症(PKU)的研究主要集中在确定PAH基因突变的位点和类型,以及如何针对其突变在基因水平给予预防、控制和修复。相信在不久的将来,人类可以在基因水平上彻底治愈苯丙酮尿症(PKU)及其他类似的基因突变导致的疾病。
-
焦点事件

-
焦点事件

-
焦点事件

-
科技前沿

-
焦点事件
-
焦点事件

-
科技前沿

-
焦点事件

-
会议会展

-
政策法规

-
焦点事件

-
技术原理

-
政策法规

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件