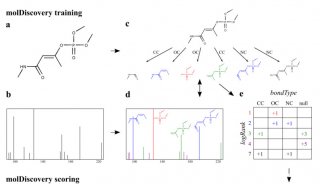
FDA强制23andMe公司个人基因组检测产品撤市
2013年11月22日,美国FDA做出令人惊讶的举措,命令基因组学公司23andMe停止上市销售其旗舰产品 PersonalGenomeService(PGS)。PGS是一种直接面向消费者销售的DNA测序产品,称这款产品“可以帮助个人及他们的医生确定他们需要留意的健康部位。”通过测定某些单核苷酸多态性的存在,PGS可预测250多种疾病的风险及通过调查研究外推个人的健康情况。2013年12月6 日,23andMe留意到FDA的警告信,然后停止提供与健康有关的基因检测。
PGS是一款以患者为中心的工具,它能提供消费者大量的遗传信息。例如,它能提供与24种遗传障碍有关的58种基因突变信息。PGS也预测200多种其它健康症状风险,如糖尿病、脂泻病和不安腿综合征。尽管这种信息可以授予患者,但它在临床实践中的作用尚不明确。
例如,FDA批准的华法林抗凝血药标签建议,如果能获得患者基因型可以按遗传学剂量用药。然而,最近的临床试验对系统基因分型产生怀疑。PGS的其它作用,如预测不安腿综合征的风险是基于初步的证据。PGS将所有这些基因检测捆绑到一个产品当中,通过互联网、广播和电视直接销售给消费者,不允许按选择的检测项目定货。这款产品已卖给了50多万个人,但它从未获得FDA的批准。
包括用于诊断的如PGS在内的医疗器械由 FDA进行监管,通常需要通过两种监管途径中的一种进行审评。医疗器械分为I、II、III类。根据FDA的警告信,23andMe在2012年提交了几项510(k)许可申请,称该公司正寻求批准PGS为II类医疗器械,这时PGS已在消费者中使用了5年。FDA因公司不能提供要求的信息,几项 510(k)申请被FDA拒绝。警告信指出PGS实质上不等同于现有的器械。
由于23andMe能够在未获得监管批准的情况下上市销售PGS,所以其测序程序及对疾病风险的预测均缺乏数据的准确性。PGS获得准确的DNA序列需要多项步骤,包括恰当的样本收集、运输和存储,以及精确测序。任何一个步骤均可出现差错,250多种病症就放大了不准确结果的可能性。数据证明测序的准确性要以FDA批准作为先决条件。对于划分为II类医疗器械的MiSeqDx,FDA审查了其有效性和质量数据,证明这款产品能准确测序各种特征明显的基因。
即使能确定PGS可以给出正确的DNA序列,其对疾病风险预测的准确性也必须进行评价,因为它们是基于临床疾病有关的特定基因序列研究外推之后得到的结果。使用PGS获得不准确结果的潜在影响通过其直接向消费者供应而被放大。如FDA建议患者服用华法林可根据PGS提供的华法林代谢信息独自调整用药剂量。在缺乏数据的情况下,要确定FDA的担忧是否有根据及PGS对独立患者使用是否适合是不可能的。此外,从PGS获得信息的潜在临床收益仍不明确。与可以证实一项肺炎诊断的胸部造影不一样,基因检测旨在预测未来潜在的疾病风险。虽然一些不确定性可以接受,但不准确的诊断检测确能带来风险,并一直与长期的心理后遗症及高昂的健康护理成本有关。
PGS是一款颠覆性产品案例,挑战了FDA现有的监管框架。由于缺乏判定的数据基础,所以FDA采用了不同寻常的行动,在很大程度上根据PGS可能导致疾病风险的不准确预测及解释的理论风险而强制这款产品撤市。明显强调理论伤害是很有意义的,因为有史以来FDA一直是一个被动的机构,在行动之前等待积累实际的安全性证据。
如果有其它数据可以阐述PGS的准确性,包括其测序程序和风险预测,可以促使这款产品重新上市。当PGS的测序程序变得准确明晰时,这款产品可能会被允许在医师的监督下使用。直接面向消费者及诊室为基础的基因组检测将会在医师发挥重要作用的临床护理中变得更加常见,其中检测结果要由医师进行分析。但只有当这款产品对疾病风险预测被证实是准确的及能在不需要医疗监督下安全使用时才能被允许直接面向消费者销售。
-
科技前沿

-
产品技术

-
焦点事件

-
项目成果
-
科技前沿

-
焦点事件

-
焦点事件

-
科技前沿

-
项目成果
-
项目成果

-
会议会展

-
焦点事件

-
焦点事件

-
科技前沿
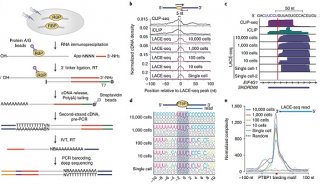
-
科技前沿

-
焦点事件

-
科技前沿
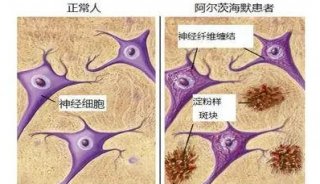
-
会议会展

-
综述

-
焦点事件
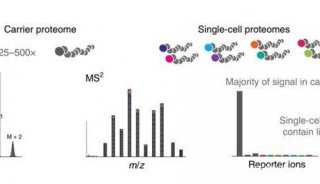
-
焦点事件

-
焦点事件

-
技术原理

-
焦点事件

-
项目成果

-
项目成果