Nat Comm| 张强锋组利用人工智能算法分析单细胞ATAC-seq数据
近年来,深度学习等人工智能技术在图像识别,自然语言处理等领域取得了令人瞩目的成就。人工智能技术应用于生命科学,对生物信息多个领域产生了重大的影响。深度学习算法借助于生命科学大数据的飞速发展,从海量大数据中自动高效地提取特征进行学习,极大超越了依赖人工提取特征的传统人工智能算法,在医疗图像处理、结构预测、药物研发、以及基因组数据分析等诸多领域大显身手。在生命组学的前沿单细胞RNA测序领域,深度学习等人工智能算法也逐渐得到应用,并取得不错的结果。然而深度学习应用于单细胞测序数据分析,面临着数据稀疏性、维度灾难、以及难以解释等诸多挑战。
染色质开放区域是基因组编码生命信息的窗口,在染色体开放区域上包含了很多重要的转录因子结合位点和基因调控的基因组元件【1】。ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing)技术通过Tn5 DNA转座酶在开放染色质两端插入测序接头来标记该区域,从而获得开放染色质的位置、转录因子结合位点、核小体的调控区域以及染色质状态等信息【2】。单细胞测序技术凭借着可以完美地解决微量细胞、样本稀少、细胞异质性等问题的优势,已经广泛应用于发育、肿瘤、免疫、神经等领域。为进一步获得“高分辨率”的单细胞精度的染色质开放图谱,近几年科学家们发展了单细胞ATAC-seq 技术,通过分析不同细胞个体及亚群的染色质开放图谱差异来揭示细胞异质性的调控机制【3, 4】。
单细胞ATAC-seq数据具有以下几个特点:高维度,即可以检测到的可能开放区域高达几十万;二值性,即每个开放区域在二倍体基因组上通常只有两个拷贝,造成数据接近于二值化;极端稀疏性,由于细胞异质性以及技术原因,导致对每一个单细胞都有大量开发区域没有信号。这些数据特征给单细胞异质性分析带来了巨大的挑战【3】。传统的降维和可视化方法像PCA和t-SNE,以及基于样本距离聚类的方法都无法在单细胞ATAC-seq数据上取得很好的效果。另外,由于单细胞ATAC-seq相比于单细胞RNA-seq数据,具有二值化和更加稀疏的特性,直接应用单细胞RNA-seq的软件和方法来分析单细胞ATAC-seq数据通常效果也不太理想。
2019年10月8日,来自清华大学生命科学学院的张强锋教授团队在Nature Communications上在线发表了题为SCALE method for single-cell ATAC-seq analysis via latent feature extraction 的工作,通过深度学习的方法提取数据低维的隐层特征,来对单细胞ATAC-seq 数据进行聚类、可视化、缺失值填补、降噪、以及下游生物信息挖掘,有效地解决了数据高维度稀疏性的问题。

SCALE 的算法框架结合了变分自编码器(VAE)和高斯混合模型(GMM)。变分自编码器是一类无监督的生成模型,可以用来进行数据的特征提取;高斯混合模型是多个高斯分布的线性组合。SCALE结合两者来拟合多模态的单细胞ATAC-seq数据分布(图1)。
图1. SCALE的模型框架
相同类别细胞的染色质开放图谱具有相似的模式(pattern),但在有着大量缺失值的高维染色质开放图谱空间中难以识别这些模式,更难以做基于这些模式的聚类分析。若用一些低维的抽象特征表示细胞,每一维的特征代表一定的模式,在这样低维的特征空间缺失值会大大降低,相似的细胞可以更容易地聚集在一起并且彼此共享信息。SCALE充分利用这一特点,通过变分自编码器的编码器(Encoder)提取满足高斯混合模型的低维隐层特征,使得不同维度之间具有低耦合性,分别表征不同细胞类别的模式,而相同类别的细胞彼此之间共享高斯混合模型的参数,从而实现共享信息来达到相互弥补缺失信息的效果,最终通过解码器(Decoder)还原出弥补缺失值后的增强数据(Enhanced data)。在此基础上,SCALE对这些可以表征细胞类别的隐层特征进行聚类(Clustering)、可视化(Visualization),和更多数据分析。
通过SCALE增强的数据可以很好用于下游生物信息挖掘。在小鼠的前脑Forebrain数据集上,相比于原始数据,在SCALE增强的数据上,通过motif 富集软件chromVAR【5】分析,可以发现更多的、特异性更显著的转录因子(图2)。比如对macroglia细胞的激活至关重要转录因子Mafb,只在SCALE增强的数据上被发现在macroglia(MG)细胞中显著富集;又比如在excitatory neurons(EX)细胞中扮演重要调控角色的转录因子Neurog1,在excitatory neurons细胞的富集效果, SCALE增强的数据显示了相比于原始数据更显著的特异性。
图2. 在SCALE的增强的数据上发现更多、特异性更显著的转录因子
此外,SCALE还具有消除批次效应的潜力。批次效应是指由于实验技术、器材等非生物样本原因导致的数据上存在整体差异,在单细胞测序的数据分析中是一个重大挑战。研究者发现SCALE提取的特征可以捕捉到批次效应,通过将这些与批次效应相关的特征去除之后,可以消除批次差异,只保留生物学上的差异(图3)。比如小鼠breast tumor 数据集,是由两盘Epcam+细胞和两盘CD45+细胞组成,SCALE可以很好地将两类细胞分开。然而分析发现,可视化的细胞群体中存在一些Plate相关的偏差。通过进一步分析SCALE提取的特征,发现有一些维度的特征与Plate相关,而另外一些维度的特征则是Plate无关的,只表征细胞类型。在后续分析中,只要剔除Plate相关特性,可以起到消除批次效应的效果。
图3. SCALE消除批次效应
SCALE目前在GitHub上开源共享(https://github.com/jsxlei/SCALE),普通用户可以通过一键安装免费便捷地使用SCALE,商业用户需联系作者获取使用授权。
原文链接:
https://www.nature.com/articles/s41467-019-12630-7
参考文献
1.Tsompana, M. and M.J. Buck, Chromatin accessibility: a window into the genome. Epigenetics & Chromatin, 2014. 7(1): p. 33.
2.Buenrostro, J.D., et al., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods, 2013. 10(12): p. 1213-8.
3.Buenrostro, J.D., et al., Single-cell chromatin accessibility reveals principles of regulatory variation. Nature, 2015. 523(7561): p. 486-90.
4.Cusanovich, D.A., et al., Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science, 2015. 348(6237): p. 910-4.
5.Schep, A.N., et al., chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods, 2017. 14(10): p. 975-978.
专家点评
张学工(清华大学自动化系/北京信息科学与技术国家研究中心 教授)
世界上没有两片相同的树叶,对于多细胞生物来讲,同样也没有两个相同的细胞。近年来单细胞测序技术的发展,使得揭示细胞异质性机制成为可能,大大推动了肿瘤、免疫、发育等诸多学科的研究。而最新的单细胞ATAC-seq技术更是促进了科学家们对不同细胞类型在基因调控层面的异质性有了更深的认识。如果说单细胞RNA-seq揭示的是细胞的分子表型特征,那么单细胞ATAC-seq揭示的则是基因表达调控机制的信息。随着单细胞ATAC-seq的大量应用,迫切需要一个能高效分析单细胞ATAC-seq数据的软件。
张强锋教授实验室如及时雨般提出SCALE工作,很好地解决了单细胞ATAC-seq数据的高维度、高稀疏性等问题。SCALE利用神经网络变分自编码器学习得到低维特征,同时利用高斯混合模型对数据分布的结构进行拟合,二者巧妙结合,形成了非常好的特征提取框架。SCALE将高维空间中存在大量缺失值的原始数据转化到有共享信息的低维空间,并让每一特征维度表征ATAC-seq数据的一个可解释性模式,是一个非常巧妙的构思,值得借鉴。总的来说,SCALE充分利用数据的分布特点,结合深度神经网络和高斯混合模型,为研究人员提供了一个非常强大的分析单细胞ATAC-seq数据的工具,为准确挖掘染色质开放图谱背后的表观遗传调控机制提供了有力助手。
专家点评
邢毅(宾夕法尼亚大学/费城儿童医院 教授)
近年来,深度学习的发展对整个生物信息领域产生了重大的推动作用,在许多领域上大放异彩,尤其是在拥有海量数据的基因组学数据分析上取得了令人惊艳的成效。单细胞ATAC-seq数据存在着维度灾难和大量信息缺失等问题,如何从数据中将不同亚型的细胞精准归类并从中挖掘出细胞特异性的调控元件非常具有挑战。
张强锋团队提出的SCALE工作,成功地解决了上述难题,给从事单细胞ATAC-seq数据分析的研究人员提供了强大的工具。SCALE充分利用数据本身的分布结构,设计模型提取隐层特征,使其不但能够拟合数据分布,同时表征具有生物学意义的数据模式,在此基础上围绕提取的特征展开不同层面的深度分析。SCALE在可视化、聚类、数据缺失值填补等结果上的都取得相当不错的效果。研究工作在各方面对SCALE的功能和特性进行了大量深入讨论,完成度非常高。期待SCALE作为一个功能强大的单细胞ATAC-seq数据分析软件,可以推动单细胞ATAC-seq测序更广泛的应用。
-
项目成果

-
精英视角

-
并购

-
会议会展

-
项目成果

-
企业风采

-
项目成果

-
企业风采

-
焦点事件

-
项目成果

-
科技前沿

-
科技前沿

-
招标采购

-
焦点事件

-
招标采购

-
项目成果

-
招标采购

-
产品技术

-
科技前沿

-
项目成果
-
焦点事件

-
科技前沿
-
科技前沿

-
项目成果
-
焦点事件

-
焦点事件

-
科技前沿

-
科技前沿
-
焦点事件

-
焦点事件

-
科技前沿
-
项目成果
-
科技前沿
-
科技前沿
-
科技前沿

-
科技前沿

-
技术原理
-
企业风采

-
焦点事件

-
科技前沿

-
科技前沿

-
科技前沿

-
技术原理
-
焦点事件

-
项目成果
-
项目成果

-
焦点事件

-
焦点事件

-
项目成果

-
项目成果
-
焦点事件

-
科技前沿
-
企业风采

-
项目成果
-
项目成果
-
焦点事件
-
项目成果
-
项目成果
-
焦点事件

-
项目成果
-
项目成果
-
项目成果
-
项目成果
-
焦点事件
-
项目成果
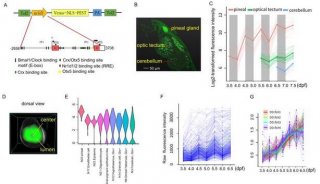
-
焦点事件
-
焦点事件
-
项目成果
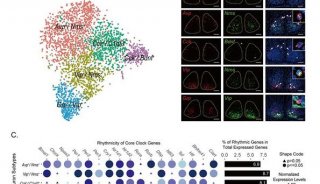
-
项目成果
-
焦点事件

-
焦点事件
-
项目成果

-
焦点事件

-
项目成果

-
项目成果

-
项目成果

-
投融资

-
焦点事件

-
焦点事件

-
项目成果

-
项目成果

-
项目成果
-
项目成果
-
项目成果
-
科技前沿

-
项目成果
-
科技前沿
-
焦点事件
-
焦点事件

-
科技前沿

-
精英视角

-
科技前沿