化合物活性预测相关知识问答
Docking相关
A:新手想问一下,docking结果怎么判断小分子和蛋白产生了结合呀,形成了疏水键或氢键吗?
B:打分越低越好,一般会设置一个已知的配体用来做参照,或者说用来检验参数是否合理。
A:我是选择了有配体的蛋白,处理了蛋白和分子后进行docking,得到这个结果。
B:只要比这个配体的打分低,那肯定就更有可能结合在那里呀!
E:新手想问下,配体和蛋白对接的时候怎么确定对接盒子的位置和坐标?
D:选中ligand。
B:根据前期文献调研,需要大体知道结合位点的位置。否则就得进行全局docking。
E:下载pdb的蛋白的原文献吗?全局docking感觉不准啊。
D:你得先知道ligand结合口袋。
B:不一定,比如一些突变研究会告诉你大体结合位点。
D:可以用氨基酸残基定位结合口袋。
(提示:更多干货见《如何确定对接口袋?》)
F:我有个问题,已知配体结合能-9,我的一堆配体结合能-2~-4,有意义么?结合位置一致。
殷赋科技:vina对接吗?大于 -4基本没什么意义。
F:是autodock的,我是一堆多肽混合物,都鉴定了成分,然后作为配体做的对接,基本都是-2~-4。基本就是没啥意义的?
殷赋科技:光看数值不可靠,要看结合模式正常吗?
F:结合模式怎么看呢,看哪几个参数么?
殷赋科技:可以对比原来的配体,看你的配体是不是结合在对应的口袋内,有没有充分结合在里边,还是有很大一部分暴露在溶剂中(考虑到打分这么差,结合模式应该不太好)。
你对接的是多肽,分子量应该不小,光疏水作用都不会让打分这么差,所以我怀疑结合构象有问题。
B:可以后面加一段分子动力学模拟。
F:我是chemdraw上画的结构又能量最小化的,然后在ADT上处理的,选择torsion的时候,显示结构中可旋转键大于32,我就调到32以内,不知道这么处理对不对。
殷赋科技 :多肽是几肽啊?起码也得5、6肽了吧。
F:从六肽到十肽都有。嗯,基本都是7,8了。
殷赋科技:我在群里前面说过,我们平台有多肽对接的功能,但是限制在6肽以下,就是考虑到可旋转键太多会造成对接不完全。
F:是啊,我这种情况该咋办呢?
殷赋科技:所以,你最好用专门做多肽对接的软件来做这个课题。
F:我看文献有一个跟我做的几乎一样的东西,他们也是用的autodock,哎,磨死我了。我多肽太多了,20多个。
殷赋科技:文献不一定正确,要持有怀疑态度。试下这个在线服务:http://biocomp.chem.uw.edu.pl/CABSdock/
F:那还是个顶刊,谢谢您解答。
殷赋科技:也不一定,看领域吧,看对接是扮演什么角色,如果只是陪衬,可能就不会好好做。
化合物活性预测
H:请问有没有比较全面的化合物活性预测平台?
殷赋科技:化合物活性预测,计算上就是虚拟筛选,比如用分子对接方法。比较全面指的是什么,化合物库多,还是软件方法齐全,还是其它?
H:意思是,对一个未知化合物,能不能进行反向对接,找到结合比较好的蛋白,然后预测化合物作用。
殷赋科技:有啊,@墨灵格
有此业务。徐老师课题组也有在线服务:http://www.rcdd.org.H/PTS/index。
墨灵格 :谢谢,基于机器学习的方法靶标预测(数据库ChEMBL24版,朴素贝叶斯多分类预测靶标):http://targetfishing.molcalx.com.cn/ml.html, 包含1900多个靶标,给出概率。
墨灵格:部分典型疾病的靶标预测(基于分子形状):http://targetfishing.molcalx.com.cn。
H:多谢,能多推荐几个类似的吗?
殷赋科技:其他我不了解。国外课题组应该也有的,在Google或bing上搜一下吧。
J:我手头也有分解的小分子产物。也想做一下和疾病相关的研究。@墨灵格
我要是想把我的小分子药物和某种疾病相关连。我如何预测啊。
墨灵格:你可以直接试试,就知道了,画结构就好。
J:嗯嗯。这个预测完了还要实验去验证吧。
墨灵格:没被验证之前都是预测,不算数。预测只是个提示,有很多东西都有待于进一步排除:只是提示你可以去关注这个靶标,然后要根据自己的知识从中排除不可能的。
如何选择 pdb code
K:请问一下,为什么一个靶点有很多的pdb code,应该怎么选择呢?
L:分辨率高的(编者按:resolution越小,分辨率越高,小于2最好),有配体的。
K:那里面的蛋白是同一个吗?我看了下序列好像有不同。谢谢你,以前都没有注意过分辨率,多谢提醒。L:如果做的是人,选人。
H:用uniprot看看。
I:集成(编者按:Ensemble,有的翻译为“系综”)对接(打分)是非常通用的策略,比传统的方法精度高非常多。集成策略已经很多年了,可以看看综述:Amaro, R. E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J. A.; Miao, Y.; Smith, J. C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114 (10), 2271–2278. https://doi.org/10.1016/j.bpj.2018.02.038.
K:@I
你好,集成策略可以用什么软件实现呢?对于药物设计帮助大不大呢?I:集成对接是个策略,你不需要特殊的软件,当然有的软件专门为这个策略定制有流程。
K:好的,我看看那篇综述,以前没有听过这个策略。I:分子对接本来就是用于早期发现,提高了命中率与精度,当然有帮助。
Miscellaneous
A:问下原配体打分怎么看啊,结果只有我的几个小分子的打分用配体dock一下吗?
殷赋科技:对!也可以只打分不对接,或者进行原位优化(in-situ minimization)。殷赋云平台上的方案有多种计算模式。
A:我用的好像没有只打分的功能。
殷赋科技:你是用我们的平台吗?
A:不是,用的moe。
殷赋科技:MOE好像没有这个功能,在它的help里边搜一下关键词看看:in situ、in-situ,对接再打分也好,能检验软件是否有能力预测正确。
C:请问用药效团pharmacophore ligand profile筛选化合物库时fitvalue多少以上的分子建议对接啊?
D:4分以上。
G: Vina后排名前七位的化合物,我将蛋白和小分子全部优化加电荷了,应该不存在缺少非极性氢和N价态的情况,cdocker后只有一个小分子对接上了,那我应该如何合理的解释这种现象比较好?根据Vina协议,VS hits肯定绑定到活性区域,而cdocker命中了一个化合物,其他VS命中化合物是否太大而无法放入cdocker对接的活性口袋中?
殷赋科技:(分子太大是)有可能的。用软件打开vina对接结果看看分子结合的情况。试下增大CDOCKER的口袋范围。也可以换其他对接方法试试,比如Dock6。
C:请问下载的pdb蛋白比如4bkx里面显示配体就一个醋酸分子,对接处理蛋白怎么定义活性位点啊?把醋酸选中吗还是从蛋白的active sites里面选一个?
G:看相关文献。
M:就选中配体,然后选择from current …生成活性球,半径啥可以再调。
C:这样做完就是发现蛋白质和配体对接结果不太好。
M:可以再试试从含这个蛋白的其他晶体结构中提取蛋白受体进行对接。
(提示:通常情况下,醋酸分子并非配体,只是溶剂或助溶剂而已;定义活性位点/口袋的方法可以参考《如何确定对接口袋?》)
更多资讯,请登录www.yinfotek.com 或关注微信公众号“殷赋科技”。我司建立了微信学术交流群,为生物医药领域的朋友搭建沟通交流的互动平台。想入群的朋友,请在微信公众号菜单栏输入“加群”,根据提示操作即可。
-
焦点事件

-
焦点事件

-
焦点事件

-
项目成果

-
焦点事件
-
技术原理
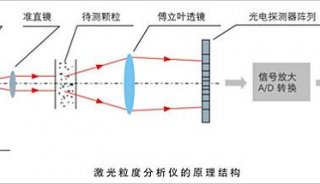
-
焦点事件

-
焦点事件

-
技术原理
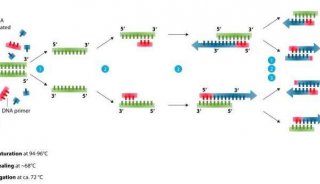
-
焦点事件

-
焦点事件