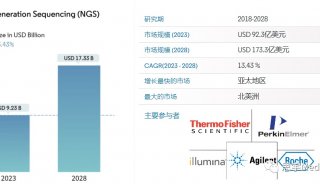
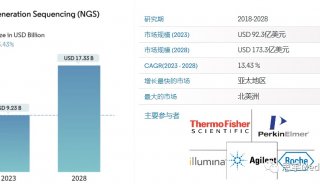
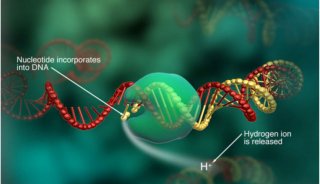
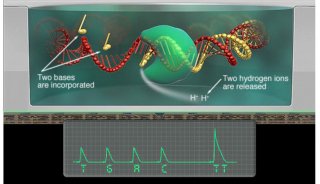
为什么deSALT技术能够突破长RNA-seq读序列比对瓶颈
由于高测序错误和复杂的基因结构,长读RNA测序读段的比对就显得非常重要。2019年12月16号,哈尔滨工业大学臧天仪、王亚东团队在Genome Biology上在线发表了题为deSALT: fast and accurate long transcriptomic read alignment with de Bruijn graph-based index的文章。研究提出了deSALT,这是一种量身定制的两遍比对方法,该方法构造基于图的比对骨架以推断外显子,并使用它们来生成剪接的参考序列以产生精确的比对。 deSALT解决了一些困难的技术问题,例如小外显子和测序错误,这些问题突破了长RNA-seq读取序列比对的瓶颈。基准测试表明,deSALT具有产生准确和均一的全长比对的更大能力。

RNA测序(RNA-seq)已成为表征转录序列的一种基本方法.它揭示了精确的基因结构,并量化了基因/转录表达在各种应用中的作用,如变异调用,RNA编辑分析和基因融合检测。然而,目前广泛应用的短消息测序技术在图书馆的准备工作中存在着有限的读取长度和系统的偏差。这些缺点限制了更精确的比对和精确的基因异构体分析,从而成为转录研究的瓶颈。
两种长读测序技术,单分子实时(SMRT)测序和纳米孔技术(ONT)产生的纳米孔测序,是转录分析中短读瓶颈的新兴和有希望的突破。它们都能够产生更长的读取,读取的平均长度和最大长度分别超过10到几十万个碱基对(BP)。利用这一优势,全长转录本可以通过单读进行测序,这对于大大提高基因异构体重建的准确性是有希望的。此外,测序过程中存在较少的系统偏倚,这也有利于基因/转录表达的定量。
deSALT方法示意图
除了它们的优点外,PacBio和OT-Read的排序错误率也比短读的高得多。对于PacBio SMRT序列,原始读段(“子读段”)的测序错误率约为10%~20%;对于ONT纳米孔测序,1D和2D(又称1D2)的测序错误率分别约为25%和12%。PacBioSMRT平台可以通过多次对循环片段进行排序来生成插入读(ROIS),从而大大减少测序错误。然而,这项技术具有较低的测序产率和减少读取长度。因此,这些高的测序误差给RNA-seq数据分析带来了新的技术挑战.读对齐可能是最受影响的,而且这种影响可能不限于读对齐本身,因为它是许多下游分析的基础。
在这里,研究人员提出了deSALT,这是一种量身定制的两遍比对方法,该方法构造基于图的比对骨架以推断外显子,并使用它们来生成剪接的参考序列以产生精确的比对。deSALT解决了一些困难的技术问题,例如小外显子和测序错误,这些问题突破了长RNA-seq读序列比对的瓶颈。基准测试表明,deSALT具有产生准确和均一的全长比对的更大能力。deSALT可在以下网址获得:https://github.com/ hitbc / deSALT。
-
科技前沿

-
科技前沿

-
政策法规
-
科技前沿

-
科技前沿

-
科技前沿

-
焦点事件

-
企业风采

-
科技前沿

-
焦点事件

-
企业风采

-
企业风采

-
科技前沿

-
企业风采

-
标准

-
产品技术

-
产品技术

-
焦点事件
-
焦点事件

-
焦点事件

-
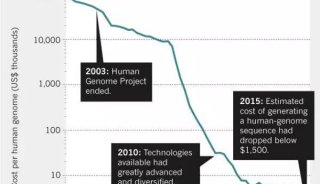
技术原理
-
技术原理

-
项目成果

-
焦点事件

-
焦点事件

-
企业风采
-
企业风采
-
焦点事件
-
焦点事件

-
综述

-
项目成果