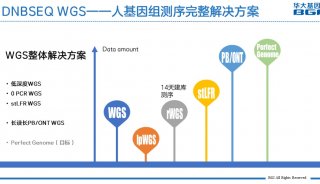
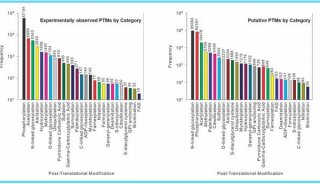
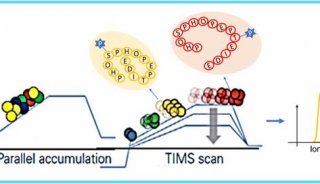
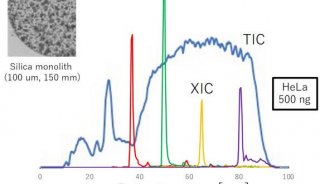
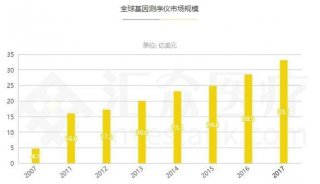
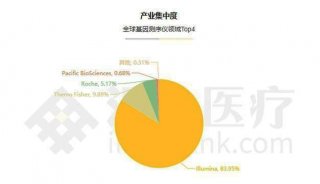
基于低深度测序的关联分析方法
由于缺少非人类生物的单倍型参考panel,研究人员很难对这类生物进行全基因组关联分析(GWAS)。为了解决这个问题,牛津大学、威康信托人类遗传学中心和加州大学洛杉矶分校的研究人员开发了一种生物信息学方法,利用低覆盖度的全基因组测序数据填充基因型。
该项研究近期发表在《Nature Genetics》上,研究小组利用2000多个测序深度仅为0.15×的封闭群小鼠的测序基因组数据和1.1万多个中国汉族人群的基因组数据证明了该方法。
发表于同一期《Nature Genetics》上的第二篇文章中,研究人员在2000个封闭群小鼠中研究了这种方法的有效性,将基因与特定性状关联了起来。
STITCH方法的开发
文章第一作者、威康信托人类遗传学中心的统计遗传学家Robert Davies将这种方法称作“通过构建单倍型进行测序填充”(Sequencing to Imputation Through Constructing Haplotypes),简称STITCH。Davies说,这种方法可以将GWAS研究的应用扩展到所有没有单倍型参考panel的物种中。他的实验室已经开始在小麦和猪中测试这种方法。
该研究小组基于需要开发出了这个方法。Davies的同事们对2000只小鼠进行了低深度测序,并尝试用已有软件对小鼠进行基因分型,但是发现现有方法都没用。于是他们开始开发一种新的方法,以在不需要额外的芯片数据或单倍型参考panel的情况下使用NGS数据。
STITCH方法基于隐马尔可夫模型,与之前开发的用于芯片数据的基因型填充方法类似。两者主要的不同在于,前者是在测序read水平分析,而应用于芯片数据的算法(例如Beagle)则是对每个SNP进行独立分析。然而利用测序分析时,多个SNP通常会出现在同一个read中。Davies说,“测序reads可以跨越4个或5个SNP。STITCH方法适应了read上的SNP并不是相互独立的这个事实。”
STITCH方法的验证
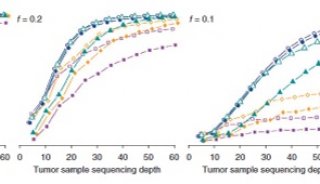
研究人员利用STITCH方法分析小鼠测序数据,估算出基因型为710万个SNP。他们将这些结果与4个测序深度为10×的小鼠基因组,以及44个之前用芯片分型得到2.1万多个SNP的小鼠基因组进行比较,验证了这些结果的准确性。
对初始结果过滤后,他们的方法与芯片结果一致性达到98%,与高深度测序的全基因组测序数据一致性达到97%。
Davies说,当他们按照“read unaware”模式运行STITCH时,准确率降低到88%,这证明考虑每个read含有多个SNP这个情况的重要性。
该小组同样还利用NGS数据测试了Beagle算法,结果发现准确率明显降低。该算法与高深度WGS数据和芯片数据的一致性分别为8%和22%。
研究人员接下来又利用1.1万多个测序深度为1.7×的中国汉族人群基因组数据测试了STITCH,将STITCH结果与72个个体的芯片分型结果、9个测序深度为10×的基因组测序结果进行比较,再次发现STITCH方法与它们的高度一致性。
第二项研究中,威康信托人类遗传学中心Jonathan Flint和Richard Mott实验室的研究人员描述了,当他们利用STITCH填充约2000个小鼠的祖先单倍型时,他们能够将一些基因与不同表型关联起来,包括与睡眠、笼内活动、惊吓反应、骨矿物质含量和创伤修复相关的基因。
作者写道,“这是第一个在没有参考panel的情况下,利用极低深度测序生成准确基因型的研究。”
STITCH方法的应用
Davies表示,STITCH可用于那些没有单倍型参考panel的非人类基因组分析中,比芯片费用更划算。据他估计,每只小鼠的低深度测序花费约60美元,而芯片分析约87美元。除此之外,测序提供的数据比芯片更多,并且不需要任何关于变异以及这些变异是如何在群体中分离的等已知信息。
Davies说,“STITCH方法需要一个高质量的参考基因组和大量样本。如果参考基因组不正确,该方法就不好用。无论如何,这种方法都是非常有前景的。”
STITCH目前免费提供给学术研究人员。Davies说,我们感兴趣这种方法接下来会如何被使用及在什么物种中使用。农业生物技术领域的研究人员可能会对它的使用特别感兴趣。
参考文献:1. Rapid genotype imputation from sequence without reference panels.Nature Genetics (2016) doi:10.1038/ng.3594
2. Genome-wide associationof multiple complex traits in outbred mice by ultra-low-coverage sequencing. NatureGenetics (2016) doi:10.1038/ng.3595
-
企业风采

-
科技前沿

-
政策法规
-
企业风采

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
会议会展

-
企业风采

-
焦点事件

-
企业风采

-
焦点事件

-
科技前沿

-
企业风采

-
科技前沿
-
企业风采

-
综述
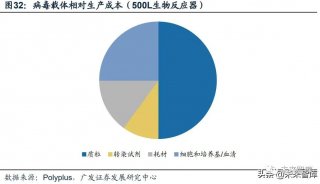
-
企业风采

-
产品技术

-
焦点事件

-
焦点事件
-
技术原理
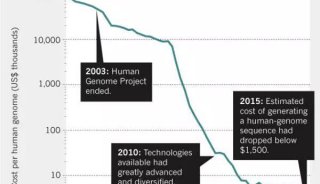
-
技术原理

-
焦点事件

-
企业风采
-
焦点事件

-
综述

-
焦点事件
-
焦点事件