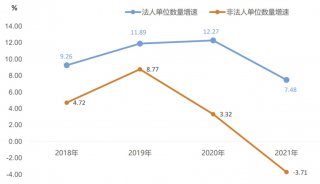
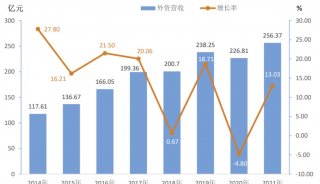
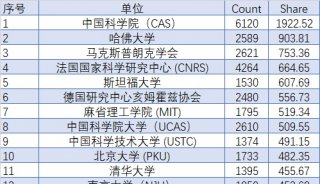
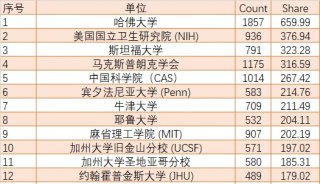
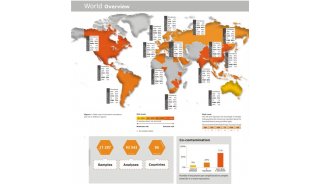
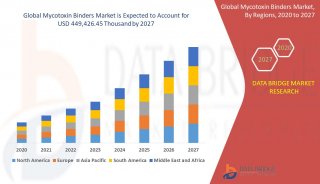
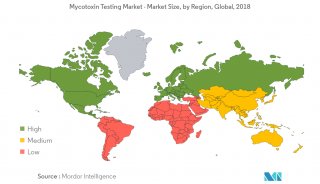
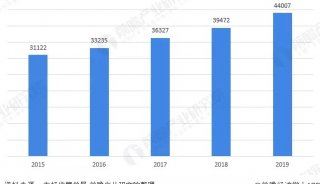
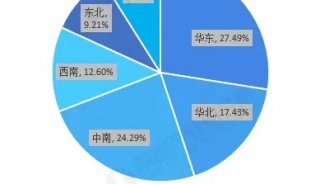
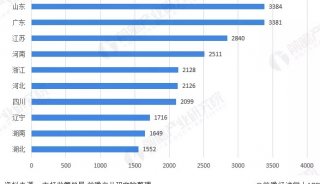
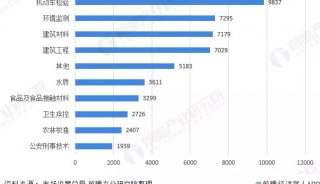
蛋白质三级机构预测-同源模型化法-1
蛋白质结构预测的生物学意义
生物信息学研究的一个主要目标是了解蛋白质序列与三维结构的关系,但是序列与结构之间的关系是非常复杂的。人们已经掌握了一些蛋白质序列与二级结构之间的关系,但是对于蛋白质序列与空间结构之间的关系了解得比较少。预测蛋白质的二级结构只是预测折叠蛋白的三维形状的第一步。一些结构不是很规则的环状区域与蛋白的二级结构单元共同堆砌成一个紧密的球状天然结构。在蛋白质折叠过程中一系列不同的力都起到了重要作用,包括静电力、氢键和范德华力;疏水作用也是影响蛋白质结构的重要因素;半胱氨酸之间共价键的形成在决定蛋白构象中也起了决定性的作用;在一类称为伴侣蛋白的特殊蛋白质作用的情况下,蛋白折叠问题变得更复杂。伴侣蛋白通过一些未预知的方式改变蛋白质的结构,但这些改变方式是很重要的。
同源模型化法
模型基础
同源模型化方法是蛋白质三维结构预测的主要方法。对蛋白质数据库PDB分析可以得到这样的结论:任何一对蛋白质,如果两者的序列等同部分超过30%
(序列比对长度大于80),则它们具有相似的三维结构,即两个蛋白质的基本折叠相同,只是在非螺旋和非折叠区域的一些细节部分有所不同。蛋白质的结构比蛋白质的序列更保守,如果两个蛋白质的氨基酸残基序列有50%相同,那么约有90%的α碳原子的位置偏差不超过3%。这是同源模型化方法在结构预测方面成功的保证。
模型的基本思想
同源模型化方法的基本思想:对于一个未知结构的蛋白质,首先通过同源分析找到一个已知结构的同源蛋白质,然后,以该蛋白质的结构为模板,为未知结构的蛋白质建立结构模型。这里的前提是必须要有一个已知结构的同源蛋白质,这个工作可以通过搜索蛋白质结构数据库来完成,如搜索PDB。同源模型化方法是目前一种比较成功的蛋白质三维结构预测方法。从上述方法介绍也可以看出,因为预测新结构是借助于已知结构的模板而进行的,选择不同的同源的蛋白质,则可能得到不同的模板,因此最终得到的预测结果并不唯一。
模型预测的基本步骤
假设待预测三维结构的目标蛋白质为U(Unknown),利用同源模型化方法建立结构模型的过程包括下述6个步骤:
1、搜索结构模型的模板(T):同源模型化方法假设两个同源的蛋白质具有相同的骨架。为待预测的蛋白质建立模型时,首先按照同源蛋白质的结构建立模板T。所谓模板是一个已知结构的蛋白质,该蛋白质的与目标蛋白质U的序列非常相似。如果找不到这样的模板,则无法运用同源模型法。
2、序列比对:将目标蛋白质U的序列与模板蛋白质序列进行比对,使U的氨基酸残基与模板蛋白质的残基匹配。比对中允许插入和删除操作。
3、建立骨架:将模板结构的坐标拷贝到目标U,仅拷贝匹配残基的坐标。在一般情况下,通过这一步建立目标蛋白质U的骨架。
4、构建目标蛋白质的侧链:可以将模板相同残基的坐标直接作为目标蛋白质的残基坐标,但是对于不完全匹配的残基,其侧链构象是不同的,需要进一步预测。侧链坐标的预测通常采用已知结构的经验数据,如ROTAMERS数据库。ROTAMERS含有所有已知结构蛋白质中的侧链取向,按下述过程来使用
ROTAMER:从数据库中提取ROTAMER分布信息,取一定长度的氨基酸片段(对于螺旋和折叠取7个残基,其它取5个残基);在U的骨架上平移等长的片段,从ROTAMER库中找出那些中心氨基酸与平移片段中心相同的片段,并且两者的局部骨架要求尽可能相同,在此基础上从数据库中取局部结构数据。
-
政策法规

-
焦点事件

-
会议会展

-
政策法规

-
项目成果

-
焦点事件

-
焦点事件

-
科技前沿

-
产品技术

-
焦点事件

-
科技前沿

-
项目成果
-
企业风采

-
科技前沿
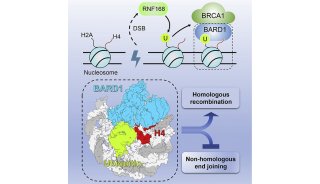
-
科技前沿

-
科技前沿
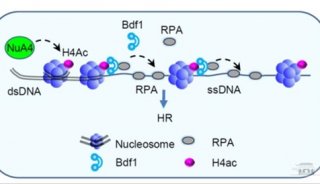
-
综述
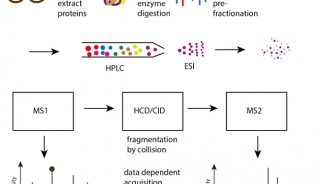
-
焦点事件
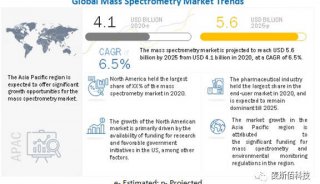
-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
焦点事件
-
焦点事件

-
焦点事件

-
项目成果
-
项目成果
-
焦点事件
-
综述