DNA测序40周年:DNA测序的过去、现在和未来(上篇)
在DNA测序过去的40年中,我们见证了诸多技术的变革和测序规模的极度增长。从几千个碱基到第一个人体基因组,乃至当前数以万计的人体和无数其它的基因组。包括作为大量分子现象的“计数器”在内,DNA测序被广泛和创造性地应用于各个领域。从长远来看,我们可以预测DNA测序技术所带来的影响将会与显微镜的使用相媲美。
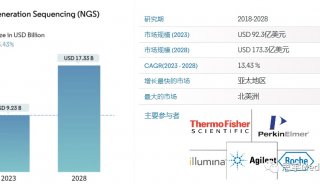
华盛顿大学基因组科学学院于十月份在国际顶级期刊《Nature》上发表了题为“DNA sequencing at 40: past, present and future”的综述文章,用以纪念DNA测序四十周年。文章主要包含4个:1.DNA测序技术发展史,2.DNA测序的应用,3.DNA测序的未来,4.DNA测序——新的显微镜。本次主要为大家带了第1部分内容的介绍(图1)
DNA测序技术发展史
DNA测序技术的发展历史很丰富,在几十年间发生了多个模式的转换。下面,我们回顾一下对生物聚合物进行测序的早期努力:电泳法DNA测序的发明和它们在人类基因组计划上的拓展,以及二代(大规模平行测序,高通量)和三代(实时,单分子)测序技术的出现。
早期测序技术
Fred Sanger把自己的科研生命贡献到了对基本序列的确定,他认为我们需要对生物分子特定化学结构的知识进行更深层次的了解。极具讽刺的是,即使是基于当前的生物高聚物进行测序技术,我们首先了解的却是蛋白和RNA。
在20世纪50年代早期,Sanger确定了第一个蛋白序列,即胰岛素,他把胰岛素断裂成两条链,解码每一条片段,然后通过两条链之间的重叠区域将它们拼接成一条完整的链。他的这个工作明确地揭示蛋白具有特定模式的氨基酸残基。随后发展的Edman降解法,通过连续降解肽链的N端残基,使蛋白测序变得更加简单。即使这些方法显得很笨重,但是到20世纪60年代末期,很多蛋白的序列已经被测定,显而易见,每个蛋白序列在物种和个人之间是不同的。
在20世纪60年代,通过相同的处理过程解决了RNA测序问题:首先用RNases将RNA片段化,紧接着通过层析和电泳技术对这些片段进行分离,然后通过连续的外切酶降解对单个片段的序列进行测定,最后通过每个片段之间的重叠区域将它们连接成完整的RNA序列。第一个测序的RNA序列,即丙氨酸tRNA,对于其1g纯净的样本,需要5个人工作3年才能确定其76个核苷酸。“指纹”技术对该过程进行了大大的简化,包括对放射性标记的RNA片段进行双向分离和可视化,然后通过最终的位置来确定RNA的大小和序列。
DNA测序技术的发明
对DNA序列进行测定的早期尝试是很麻烦的。在1968年,Wu报道了通过引物延伸法来确定lambda噬菌体粘性末端的12个碱基。在1973年,Gilbert和Maxam报道了乳糖抑制物结合位点的24个碱基,该结果是通过将该序列复制到RNA序列中,然后对RNA进行测序而得到的,该过程花费了2年,一个月测定一个碱基。
在大约1976年,两个能够在一个下午对成百上千个碱基进行测定方法的发明改变了该领域。这两个方法是:Sanger和Coulson发明的链终止法和 Maxam和Gilbert发明的化学切割法,这两种方法都是利用放射性标签到每个碱基在DNA上所处位置的距离来确定核苷酸顺序的。Sanger的方法涉及到荧光标记引物的四次DNA聚合酶扩增,每次扩增利用微量的链终止核苷酸,产生不同长度的片段。Gilbert的方法利用末端标记的DNA限制性片段,并且在四个反应中,利用化合物对特定碱基进行部分切割。对于这两种方法,利用聚丙烯酰胺凝胶电泳来测定每个碱基特异性反应产生的片段大小。在凝胶上,每个泳道包含一个碱基,通过X射线照射产生一个具有梯度的图像,通过图像可以快速读取序列,按照大小对四个泳道进行排序,从而获得每个碱基的顺序。
这些方法迅速地得到了应用。1979年Staden提出了鸟枪法测序-对随机克隆子进行测序,然后基于重叠区对序列进行拼接,在1980左右,Messing发明的单链M13噬菌体克隆载体极大地改善了该技术,并被用于基因组的de novo组装,例如早在 1982年,利用该方法测定了lambda噬菌体的基因组。1987年,Smith和Hood发明了自动的、基于荧光的Sanger测序仪,并将其用于生物系统,每天可以产生大约1000个碱基。序列数据以指数形式增长,逼近了摩尔定律,促进了中心数据库的产生(如GenBank),通过搜索工具(如BLAST),放大每条序列的值,并且形成了数据共享的思想。在1982年,超过50万的碱基已经被提交到了GenBank ;到1986年,有将近1000万的碱基 (GenBank和WGS统计数据 https://www.ncbi.nlm.nih.gov/genbank/statistics/)。
人类基因组的扩展
“层级shotgun”策略是人类基因组(HGP)计划的主力军,它把大片段的人类基因组序列克隆到细菌人工基因组上(BACs)。对来自于每个 BAC的DNA进行片段化、大小筛选和亚克隆。挑选和培养单个克隆,然后分离DNA。纯化后的DNA作为Sanger自动测序的模板,从凝胶的激光扫描图中获取信号,最后调用碱基来生成序列。事实上该过程涉及到很多独立的步骤,每一步必须很好的实施,让怀疑论者去怀疑在任何合理的成本下去足够高效地测定人类基因组吧。
的确,随着对较大基因组测序的努力已经成形,每一步测序的规模和效率都需要大大的提升。在20世纪90年代,这些想法在适应和冲刺中得以实现。值得注意的改进包括:(1)由染料标记的引物向染料标记的终止剂转变,反应由原来的四个变成了一个;(2)一个突变的T7 DNA聚合酶更容易融入染料标记的终止子;(3)线性扩增反应,极大地降低了对模板的需求,并且促进了小型化的进程;(4)磁念珠DNA纯化方法,简化了自动的焦磷酸化步骤;(5)能进行双链DNA测序的方法,该方法促进了质粒克隆的应用和随后的双端测序;(6)毛细管电泳消除了凝胶的浇注和装载,同时也简化了荧光信号的提取和翻译;(7)采用工业过程来提高效率和降低误差(如自动化、质控、标准的操作过程等等)。
潮湿的实验室协议只是挑战的一半。花费大量的努力来开发能追踪克隆、翻译和组装测序数据的软件。如,phred的发明代替了测序序列的人工编辑,它引入了可用于碱基调用的质量衡量标准,并且帮助整理出紧密相关的重复序列。然后用质量感知的方式,根据序列间的重叠区对序列进行组装,产生长度连续的序列。随着更加复杂的基因组被攻克,重复的序列越来越混杂。甚至在对一个BAC进行深度shotgun测序之后,一些序列仍然未出现,这样就导致序列的不连续,必须通过其它方法来解决这种不连续的问题。双端测序帮助将contigs连接成有缺口的scaffolds,可以通过直接测序来填补scaffolds的缺口。有些问题只能有肉眼进行解决;科学家被训练成“修理工”,去评估单克隆测序序列的组装质量。
虽说在轮廓上该过程依然稳定,20世纪90年代测序过程的快速改善导致测序成本的稳步下降,而并行计算的发展很快替代了人的决策。到2001年,少数学术基因组中心通过操作自动化生产线每天可以产生超过1000万的碱基。基因组组装软件在人类基因组计划内外都很成熟了,例如phrap、TIGR和Celera组装软件,能够处理日益复杂的基因组。每年倍增的容量使我们能够成功地完成对一些高质量基因组的测定,从Haemophilus influenza(嗜血杆菌,大约2Mb,1995年)开始,紧接着是Saccharomyces cerevisiae(酿酒酵母,大约12Mb,1996年)和Caenorhabditis elegans(秀丽隐杆线虫,大约100Mb,1998年)。人类基因组计划所测定的人类基因组,其大小是秀丽隐杆线虫的30倍,并且具有更多的重复序列,在2001年完成了对其草图基因组的绘制,然后在2004年获得了其完整的序列。在人类基因组计划实施的同时,Venter 和Celera利用全基因组shotgun策略测定了一个人的基因组(2001年)以及Drosophila melanogaster(果蝇,大约175Mb,2000)基因组。全基因组shotgun策略与人类基因组计划测序策略不同,在下面将会进行更深一步的讨论。
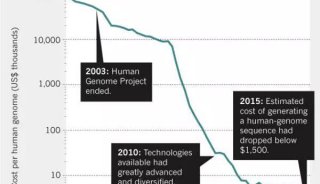
到2004年,利用仪器对含有600-700bp碱基的序列进行测定需要耗费1美元,因此再进行额外的改进也只是一个越来越边缘化实践。此外,随着人类基因组计划的完成,大规模DNA测序的未来尚不清楚。
大规模并行DNA测序技术
贯穿20世纪80年代和90年代,一些研究小组一直在探索电泳测序的替代者。虽然这些努力并未成功。直到人类基因组计划完成的10年之内,“大规模平行”或“下一代”DNA测序(NGS)几乎完全替代了Sanger测序。NGS技术在一些方法上与电泳测序显著不同,而主要的变化是多路技术。不是每个反应一个管,而是将一个复杂的DNA模板库固定到一个双向表面上,所有的模板都可以与单个试剂进行反应。该技术更不是细菌克隆,而是在体外产生大量的测序模板。最后,也不是测量片段的长度,而是测定生物化学循环(如聚合酶介导的荧光标记核苷酸)和成像(也被称为边合成边测序,SBS)。
虽然扩增并不是严格必须的(如单分子SBS),但是NGS稠密的多路技术(含有数百万到数十亿的模板)很大程度上是由体外克隆扩增获得的。最简单的方法“桥式扩增”,包括用固定在表面的引物扩增复杂的模板库,这种方式扩增得到的模板紧密成簇。作为替代,可以在乳剂中进行PCR,通过此方法获得的模板被固定在玻璃粉上,然后将玻璃粉排列到表面上进行测序。第三个方法包括在溶液中进行滚环扩增产生克隆的“纳米球” ,然后将纳米球排列在表面进行测序。
对于边合成边测序,这里有三个主要的策略。Ronaghi和Nyrèn焦磷酸测序的方法,包括离散的、分步加入每个dNTP。dNTP的加入会释放焦磷酸盐,利用萤火虫荧光素酶产生荧光。利用一个类似的方法,离子敏感场效应检测加入的天然dTNP。第二个策略是一个基于序列的方式,利用DNA连接酶的特异性将荧光寡核苷酸连接到模板上。第三个方法被证明是最持久的,包括分步的、聚合酶介导的荧光标记寡核苷酸的掺入。对于聚合酶介导的边合成边测序最重要的是:可逆终止的发展,可逆的荧光标记dNTPs,以及一个合适的工程化的聚合酶,这样对于一次循环一个模板只能合成掺入一个dNTP。通过成像确定是哪个荧光标记的dNTP被掺入到了模板,然后去掉阻碍基团和荧光标记物,将会开启下一轮扩增;这种方法是Balasubramanian和Klenerman在1998年发现的,并被用于Solexa。
第一个综合的NGS平台来自于2005年,即Solexa。Shendure、Porreca、 Mitra和Church利用该平台对大肠杆菌进行重测序,Margulies、Rothberg和454利用该平台对生殖支原体进行de novo组装,以及利用该平台对phiX174和一个人类的BAC进行重测序。这些研究证明短序列是如此的重要,并为它们的组装提供了参考基因组。在三年内,于Solexa平台上利用35bp的读长将实现对人类基因组的重测序。
在2005年,454发布了第一个商业化的NGS仪器。伴随着人类基因组计划,大规模的测序依然是少数基因组中心成立的起源。随着454和其它竞争性的仪器相继出现,个别实验室可以立即访问整个人类基因组计划时代的基因组中心。随着来自于各个方面的新方法、新结果、基因组和其它革新的出现,这种“平民化”的测序能力对基因组领域的文化和组成具有深远的影响。
与人类基因组计划期间美国应用生物系统公司的垄断不同,一些公司包括454(被罗氏收购)、Solexa(被Illumina收购)、Agencourt(被美国应用生物系统公司收购)、Helicos(由Quake成立)、Complete Genomics(由Drmanac成立)和Ion Torrent(由Rothberg成立)在激烈地竞争NGS,通过不断出现的新仪器迅速改变了以上的垄断情况,他们每年都会在硅谷的AGBT会议上发布新的仪器。在2007到2012年间,DNA测序的每个碱基的耗费下降了4个数量级。
自从2012年,测序仪器更新的速度已经变缓,各大公司之间的竞争也是一样的。454、SOLiD 和Helicos平台不再开发新的仪器,Illumina平台开始占主导优势(尽管Complete Genomics依然是一个潜在的竞争对手)。尽管如此,自从2005年NGS概念的提出开始,当考虑到我们已经走了多远的时候,还是很令人震惊的。虽然序列读长依然短于Sanger测序,只有几百个碱基,但是它的正确率超过了99.9%。花费几千美元,一个毕业生可以利用一个Illumina NovaSeq仪器在两天内对长度超过几十亿碱基的独立序列进行测定。这超过了人类基因组计划产生的大约23兆碱基的人类基因组草图40个数量级。
实时单分子测序技术
上述提到的几乎所有的平台都需要模板扩增 。然而,扩增的缺点包括复制错误、基于序列的偏好性和信息的缺失(例如,甲基化),更别说添加的时间和复杂性。在理想条件下,测序是准确且不受读长限制。为了达到这个目标,可追溯到20世纪80年代,对少数群落的探索甚至比NGS方法更基础。许多方法都已到尽头,但至少两种方法没有,这些方法就是最近兴起的再次颠覆这一领域的实时单分子测序平台。
第一种方法,由Webb和Craighead提出,由 Korlach,Turner 和Pacific Biosciences (PacBio)进一步发展,在实时测序中利用光学观察聚合酶的合成。一个零模波导孔不到光波长的一半,当模板与单个聚合酶发生反应被激发的荧光限制在小孔内。因此,只有标记荧光的核苷酸DNA链合成发出连续的信号才能被捕获到。合成的聚合酶是具有高度活性的,通常可以读取10 kb,一些reads可达到100 kb。PacBio的通量始终比NGS平台(如,Illumina公司)的最高通量低于在一个数量级。PacBio的错误率很高(约10%),但都是随机分布。PacBio的最小偏差(例如,极端GC含量的耐受)、随机误差、长读长和高覆盖度可使de novo组装更加准确,许多物种可以超越可能甚至与HGP相似。
第二种方法是nanopore测序。这个概念于20世纪80年代首次提出,基于流动离子的概念。当单链DNA分子通过狭窄的通道时,也就暴露除了测序的首链。从概念念到现实需要数十年的工作。首先,DNA通过电场在纳米孔中转运,一个核苷酸有数个电子,因此释放的信号是不足的。这些和其他挑战最终都被解决,包括加入干预酶减慢进程,提高核苷酸蛋白的鉴定,更好地分析结果信号。这种进展最近成功应用藻类的Nannopore测序中,在工业和学术中,在2005年由Bayley建立的Oxford Nanopore Techno -logies (ONT)尤其显著。ONT的测序读长与PacBio相似甚至超越PacBio的测序读长。目前获得的最长的序列达到900 kb。与其他测序仪器的主要区别在于nanopore仪器的可携带性,大小等同于USB设备,因为它们依赖于电信号的检测,而不是光学信号。一些挑战仍然存在(如,测序错误可能不是随机分布),但过程很快。
核酸测序可理想的捕获到DNA的修饰。事实上,PacBio和nanopore测序都说明了原始共价修饰的检测,如甲基化作用。单分子测序方法也开启了对RNA测序甚至蛋白质进行直接测序的可能。
1977年以后,DNA测序技术的发展突飞猛进,在我们的努力下也一直发生变化。尽管Illumina是目前测序仪器的主要供应商,但是商业市场不可能是一成不变的,其他技术也可能成功地占据重要的市场(如,de novo 组装的PacBio和便携式的ONT)。NGS和单分子方法的成本和通量都很高,也有其他的观念一直在发展,这里不做讨论(如:固态孔和电子显微检查)。所有的可能并非全部实现,但通过上例可以清晰的看出测序技术的变化走向成熟需几十年。
-
科技前沿

-
科技前沿

-
科技前沿

-
招标采购

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
焦点事件

-
标准

-
人物动向

-
并购

-
项目成果

-
企业风采

-
科技前沿

-
企业风采

-
并购

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
科技前沿

-
产品技术

-
产品技术

-
产品技术

-
技术原理
-
技术原理

-
综述

-
科技前沿
-
项目成果

-
焦点事件

-
焦点事件

-
焦点事件

-
项目成果
-
科技前沿
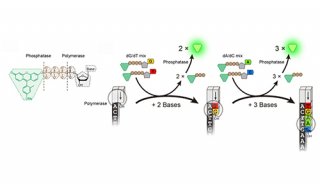
-
项目成果

-
科技前沿

-
科技前沿