
麦特绘谱生物科技(上海)有限公司

 分享至
分享至



初学者的福音—GC/MS代谢组学Q&A
点击上方蓝字关注“麦特绘谱”

GC/MS是代谢组学三大分析平台之一,因其高灵敏度、高稳定性和公共数据库定性方便等优点,成为广大科研工作者重点选择的检测平台。那么,在实际使用中我们会碰到哪些问题?针对这些问题,我们该如何解决?小编精心挑选了一些常被问到的经典问题,并给出了详细的回答。希望对您有所帮助。
1.GC/MS平台有哪些?
目前,市面上和GC串联的质谱有多种,如TOF、Quadrupole、Linear Ion Trap、Orbitrap、Sector MS以及串接质谱如Q-TOF,TQ等等。目前在代谢组学领域应用最广泛的还是GC-TOFMS平台,因为它具有高通量、高灵敏和稳定性好等优势。对于靶向定量代谢组学,则可选择GC-TQMS。
2.GC/MS分析生物样本为何要衍生化处理?有哪些衍生化的方法?
GC的流动相为气体(通常为高纯氦),这就要求被分析物必须能够气化,而生物样本中很多内源性代谢物都含有极性基团,具有沸点高、不易气化特点。衍生化能够降低这些代谢物的沸点,增加它们的热稳定性,以便分析能够顺利进行。衍生化方法及试剂种类繁多,根据不同的分析目标,需选择合适的衍生化方法。如分析脂肪酸,我们可以采用甲酯化衍生。在GC/MS代谢平台上,最常用的衍生化方法是硅烷化衍生,因为它的广谱高效。在进行硅烷化衍生之前,还需添加甲氧胺吡啶溶液,以封闭羰基(针对α-酮酸和糖类,保护作用),减少衍生化副产物的生成。

3.GC/MS能检测到哪些类别的代谢物?
衍生化极大地拓展了GC/MS的检测分析范围。GC/MS能检测到有机酸、氨基酸、单糖、糖醇、胺类、脂肪酸、吲哚、单甘油酯、核苷、二糖、生育酚、甾醇和胆汁酸等代谢物。
4.保留指数是什么?其目的是什么?
保留指数(Retention Index)又称科瓦茨(kovats)指数,是重要的定性指标。它需要采用一系列保留指数基准物质(如脂肪酸甲酯和正构烷烃)作为参考,将保留时间转换为指数。相比于保留时间,保留指数的特点是它只和色谱柱类型有关,而和其他仪器参数无关。例如,针对不同的生物样品,我们可能需要不同的升温程序,以达到最佳的色谱分离。此时,代谢物的保留时间就会发生改变,无法和质谱库中的标准时间进行匹配。再比如,色谱柱使用较长时间后,柱前端容易被严重污染,从而影响柱效。我们一般会将柱前端截断30-50公分(更好的方法是色谱柱前添加保护芯片)以恢复柱效。此时,所有代谢物的保留时间都会提前。保留指数不会受到这些实验条件的影响,依然可以用于准确定性。

5.GC/MS如何提高定性准确度?
GC/MS的定性有双重标准,一是上文提到的保留指数,二是质谱信息的相似度(similarity)。不同于LC/MS的软电离源,GC/MS使用的EI源属于硬电离,能量较强且稳定,在将代谢物分子电离后,分子离子峰进一步碎裂产生丰富的碎片离子。因此,我们很难在GC/MS上看到分子离子峰。GC/MS提供的是代谢物的“指纹图谱”,将仪器得到的实际指纹图谱和质谱库(如NIST库)进行比对后,我们将会得到一个叫相似度的指标。保留指数和相似度都必须满足设定的阈值,软件才会给出色谱峰的定性结果。最后采用标准品质谱信息进行完全比对定性,也就是说只有经过标准品比对,代谢物才是被百分之百定性。
6.什么是解卷积?
我们平常看到的总离子流色谱图(TIC)其实是软件根据质谱采集到的一个个数据点拟合出来的。每一个数据点背后就有一张质谱图。当两个或多个色谱峰没有分离开而共流出时,质谱采集到的数据点就是一张混杂的质谱图,包含了多个组分的碎片信息。如果直接用于定性分析会导致物质相似度的降低和组分的丢失。解卷积(Deconvolution)就是利用数学算法将色谱未分离的组分重新解析开,还原它们最真实的质谱信息。需要指出的是,解卷积不是万能的,好的色谱分离依然十分重要。

7.代谢物的峰面积是如何计算的?
软件会对原始质谱数据做基线计算、平滑、峰查找和解卷积。在解卷积之后,软件会考察代谢物所有碎片的信噪比、碎片提取离子流图(EIC)的对称性以及碎片色谱峰的纯度(共流出干扰的程度),最终自动挑选出一个最优的定量离子(Quant Mass)。最终的峰面积是对定量离子进行积分所得。
8.为什么有的代谢物会出不止一个峰?
怎么处理?
这主要是由于衍生化反应造成的。当代谢物有多个活泼氢时,会产生三甲基硅烷基(TMS)取代数目不同的衍生产物。如甘氨酸会生成2TMS和3TMS取代的衍生物。即使衍生试剂过量,也很难保证不同TMS取代个数的产物比例会保持一致。因此,通常的做法是将同一个代谢物的所有衍生产物的面积进行加和。我公司自主开发的XploreMET软件会自动完成这一步骤。

9.GC/MS平台数据处理软件有哪些?
在进行统计分析之前,我们需要将仪器采集的质谱数据转换为分析软件可读的数据矩阵。这其中涉及的数据处理包括基线矫正、去噪、平滑、解卷积、积分、定性、保留时间矫正和峰匹配(peak alignment)过程,最终输出一张包含样本、代谢物、保留时间和峰面积等信息的表格。商用处理软件有我公司的XploreMET、LECO公司的ChromaTOF和Spectral Works公司的 AnalyzerPro。免费的软件有Scripps研究所的XCMS、开源的MZmine 2和日本理化研究所Hiroshi Tsugawa博士主开发的MS-Dial。在使用之前,你需要将原始质谱数据转换为通用的数据格式如netCDF和mzML,或软件要求的其他格式。

10.如何获得高质量GC/MS代谢组学数据?
实验设计先不谈,在仪器分析阶段,若想获得高质量数据,有以下几个方面需要注意。
1)前处理方法的一致性。不同实验人员操作方式不尽相同。因此,一个项目应当只由一个人负责前处理。推荐使用自动衍生设备,以减少人为的误差。
2)检测之前需确保仪器处于最佳工作状态。仪器的控制软件都有系统自检功能,可以快速便捷地核查质谱仪器的状态。但更重要的是气相色谱状态,推荐使用一组混标作为仪器质控。在每一个项目开始之前,先进行质控样检测,确认气相色谱的分离度和质谱的整体响应。
3)后期数据矫正,特别是对于大样本项目。我们可以在检测序列中加入随行质控样本(每个样品取少量后混合),在后期数据处理时,使用质控样本结合如LOESS之类的算法对整体数据进行矫正。
看到这,相信您对于GC/MS代谢组学平台的认识又进一步。如有其他问题,也欢迎在我们的文章下方留言,我们将会进一步进行整理解答。
微信公众号:麦特绘谱
Tel:400-867-2686
Email : admin@metaboprofile.com
Web: www.metaboprofile.com

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
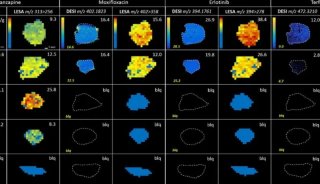
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
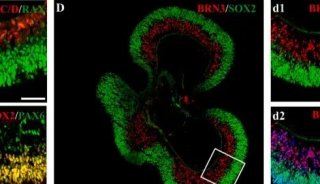
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章
-
微信文章
-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章
-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

-
微信文章

