DNA转化实验指导-2
1B. Cloning
1. A caveat on dephosphorylation: the most common reason for failure to obtain colonies is a result of adding too much BAP or CIP to the vector prep. These enzyme preparations are difficult to purify and are frequently contaminated with exonucleases and phosphodiesterases. BAP and CIP are routinely used at elevated temperatures because the contaminants are less active at high temperatures. Excess enzyme will introduce contaminating exonucleases and nibble away restriction overhangs. In addition, BAP is more heat-resistant and is difficult to completely inactivate. Precise determination of the concentration of 5’ ends is required. Calculate the exact number of ends: 2 x (g of DNA)/[size in bp x (660 Da/bp) = moles of ends of a double-stranded DNA. Use exactly enough BAP or CIP for the number of ends. This usually requires a significant dilution of the stock. Excess CIP is also reported to inhibit complete dephosphorylation. Because the byproducts of the reaction inhibit the reaction itself (dephosphorylation generally only proceeds to 95% completion), I then clean up the DNA by adding 10x stop mix consisting of 200 mM EGTA, pH 8.3, 10x TNE100 (1x TE containing 100 mM NaCl) and 10% SDS and heat inactivating at 56-68°C for 15-45 minutes; followed by phenol:chloroform extraction and ethanol precipitation. Then I repeat the entire CIP protocol all over again to dephosphorylate the residual molecules that failed to dephosphorylate the first time. This has proven to be infinitely safer than using too much enzyme. SAP is thermally sensitive and must be used at 37°C, but lacks the contamination problems of BAP and CIP. Generally, one unit of SAP is required for the complete removal of terminal phosphates from 100 pmol of 5’ ends of DNA. I have switched to the exclusive use of SAP, but supply these notes on the use of BAP and CIP for those who wish to use these enzymes.
2. Vector prep: combine 25 µg of vector (for instance, 25 mg of pUC9 equals 14.4 pmol; enough for 28 ligations using 0.5 pmoles per ligation reaction) with 20 µL of 10x KGB, 80 units @ of BamH I and Hind III, 3 units of Shrimp Alkaline Phosphatase and Type I water to a final volume of 200 µL. Incubate at 37°C for 3.25 hours. Small scale reactions using single restriction enzymes should be performed and analyzed with uncut plasmid DNA and 5 µL of the vector digest on an analytical 0.8% agarose gel to confirm that both restriction enzymes are performing as expected. Bromphenol blue runs at 800-1000 bp on an 0.8% gel. You will need to run the samples far enough to distinguish single-cut 2665 bp pUC9 from double-cut 2645 bp vector. Stain agarose gels in 1.0 mg/mL ethidium bromide or 1x SYBR Gold (Molecular Probes) for 15 minutes (no destain necessary) and visualize on a Fuji ImageReader (Fuji Medical) or photograph the gel on a UV-transilluminator (Fotodyne, Hartland, WI) with Type 667 film (Polaroid, Cambridge, MA). A typical EtBr exposure is f/5.6 for 1 1/2 sec with Kodak type 15 and type 22 filters. Rinse the transilluminator filter with water and discard gloves, KimWipes and the gel as ethidium bromide waste. Note that this is an analytical gel only.
3. It may be helpful to purify the vector by spin column chromatography or 30K MWCO cartridge to eliminate the polylinker fragment. It is not necessary to gel-purify the vector if the only cuts are in the polylinker cassette. Should you desire to do so in order to feel better, hand stain the gel in the dark with 10 mL of 1 mg/mL stain rather than use a staining bath container that is almost certainly contaminated with other plasmid DNA as well as nucleases from contaminating bacteria and molds. DNase activity in the solution can make it difficult to isolate clonable DNA from gels stained in contaminated baths.
4. Insert prep: combine 14.4 pmol of insert (PCR product or plasmid-derived) with 20 µL of 10x KGB, 80 units @ of BamH I and Hind III and Type I water to a final volume of 200 µL. Incubate at 37°C for 3.25 hours. Small scale 20 mL reactions using single restriction enzymes should be performed and analyzed with uncut insert and 5 µL of the insert digest on an analytical native 12% acrylamide gel with 20/100 bp markers (GenSura) to prove that both restriction enzymes are performing as expected. Run the gel using constant watts appropriate for the gel box. You will need to run the samples far enough to distinguish single-cut insert from double-cut insert. Stain the gel in ethidium bromide or SYBR Gold for 10 minutes (no destain) and photograph the gel on a UV-transilluminator. If ANY primer dimer contaminates the PCR reaction, the PCR fragment of correct size MUST be purified an a native 8% polyacrylamide gel prior to cloning. Passively elute or crush and soak the band of interest in 300 mM sodium acetate. Gel slices from 1-cm wells are soaked in 330 µL of NaOAc in a siliconized microfuge tube, while slices from a 1-inch are soaked in 1000 µL. Place tubes on a room temperature rotator for at least 4 hours or overnight. Ethanol precipitate the eluted DNA with Linear PolyAcrylamide (LPA) carrier. Digestion of the PCR fragment will typically generate 2 smaller bands equivalent to single- and double-cut fragments. As long as there is no uncut fragment visible on an ethidium bromide stained native polyacrylamide gel, the material is suitable for cloning.
5. If restriction digestion of the insert or vector results in multiple DNA fragments, then gel-purification is mandatory.
6. While the analytical gels are being run, clean up both the insert and vector by extracting twice with Tris-buffered phenol:chloroform, once with chloroform and then ethanol precipitate with LPA carrier. Do not use tRNA carrier, since it will inhibit the ligation. If either of the preparations require further restriction digestion, this step is much more efficient when the DNA is pre-cleaned. Repeat the set-up as described above and cut for approximately one hour. If one of the enzymes is not performing, consider switching to a new lot.
7. Ligation: mix 0.5 pmol of BamH I/Hind II-cut dephosphorylated pUC vector with 5 pmol ofBamH I/Hind III-cut insert, 2 µL of 5x TALB, 1 µL of 100 mM DTT, 1 Weiss Unit of T4 DNA ligase and Type I water in a final volume of 20 µL. Incubate the reaction at 14°C for at least 3 hours or overnight. Heat inactivate the ligase at 65°C for 5 minutes. Controls consisting of 1) the reaction minus the insert and ligase, and 2) the reaction minus the insert should be run the first time a batch of BamH I/Hind II-cut dephosphorylated pUC vector is used.
8. When bi-directional insertion of the DNA fragment into the vector is desired, in order to reduce toxic effects of the insert, the Sma I site or EcoR V (or Eco32I) site (depending upon the plasmid) may be used to linearize the vector.
1C. Notes on Cloning
1. This protocol, as written, is designed for PCR fragment inserts of 80 to 100 bp in size. For other insert sizes, the suggested insert to vector ratios are as follows: fragments < 1 kb uses a 2:1 or 3:1 ratio, inserts that are within 1 kb of the vector use a 1:1 ratio, while larger inserts reverse the ratio to 1 part insert:2 parts vector.
2. This protocol uses 0.5 pmols (866 ng) of pUC vector in a 20 mL ligation reaction for a final vector concentration of 43 ng/mL. While the quantity of vector can be scaled down to as little as 30 fmols (52 ng) of nucleic acid, the larger amount avoids problems that can arise from inherent errors in spectrophotometer measurements of nucleic acid concentrations or loss of material during purification.
3. PCR products should be cleaned by 30K MWCO cartridge with a 300 mL rinse with Type I water. This will remove most, but not all, of the residual dNTPs. Resuspend the PCR product in 30 mL of water and transfer to a siliconized microfuge tube. Determine the concentration spectrophotometrically and calculate the micromolar concentration. For an accurate concentration determination of small fragments, paste the sequence into the IDT (www.idtdna.com): Customer Service: Oligo Analyzer program to obtain the extinction coefficient. Extinction coefficient calculations are significantly more accurate than calculations using the assumptions of 1 A260 = 20 - 50 mg/mL Divide the A260-A320 value by the extinction coefficient and multiply by the dilution factor and 1x 106 to obtain the µM concentration. If the yield of PCR product is greater than the concentration of primers used in the PCR reaction, assume 100% conversion to product and 85% recovery from the 30K cartridge and divide the primer concentration by the volume of sample to obtain a crude estimate of the DNA concentration.
4. If the analytical gel indicates that the PCR product is composed of a single band, Centrex 30K purification and ethanol precipitation may be sufficient. The PCR product should be carefully analyzed by analytical gel. If the slightest trace of any primer dimer contaminates the reaction, the PCR fragment of correct size must be purified on a native polyacrylamide gel prior to cloning.
5. Because there are usually 10-20 pmol of dsDNA in the restriction digest that may contain 1013 recognition sites, modifications of normal digestion conditions are required. If timepoint analysis shows that 80 units of enzyme in a 150 µL restriction reaction for 3.25 hours are required to completely digest 25 µg of pUC vector (equivalent to 14.4 pmol) withBamH I and HindIII, then 14.4 pmol of PCR fragment will require the same digestion conditions.
-
政策法规

-
项目成果

-
标准
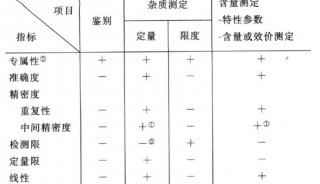
-
标准
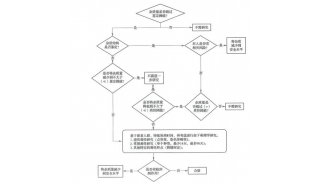
-
政策法规
-
焦点事件

-
科技前沿
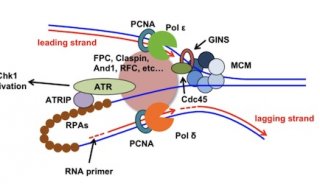
-
焦点事件

-
焦点事件

-
技术原理

-
实验室动态

-
焦点事件

-
焦点事件

-
企业风采